Center for World Health, Department of Neurosurgery, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA
Correspondence Address: Jorge Lazareff Center for World Health, Department of Neurosurgery, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA
There are two preferred ways of sharing medical information. One is data centered and for very good reasons has dominated our epistemology for almost a century. The other is empirical, based on a motley collection of hunch and experience. Both ways complement each other, both are important for our growth as physicians and surgeons dedicated to walk with our patients to their recovery.
For some reason that I am not able to explain the wonderful and experienced colleagues from Latin America and Africa are shy about telling their experience and opinion. Perhaps it is because in Latin America academic and professional promotions do not depend on the number of peer reviewed publications. In any case we at Surgical Neurology International feel that those voices need to be heard. Thus, we have launched this series that has been called “How I do it”.
We asked four Pediatric Neurosurgeons from Latin America and asked them to tell us about their approach to a prevalent condition neglected by the designers of public health strategies, neural tube defects.
The original text is in Spanish accompanied by an English translation that while short in nuances manages to be loyal to the intentions of the authors.
The papers are short, it could not be otherwise, and the authors have almost never published before. But the papers are dense in technical insight. In all the papers the reader will hear the undercurrent of devotion to the most forgotten of the patients, the malformed born in a low and middle-income country.
I praise James Ausman, M.D. the editor for accepting, supporting and encouraging this initiative. As I ready the papers for submission I recognize my shortcomings in polishing the message of my colleagues. And while coming contributions on “How I do it” will better the previous, this first series has the charm and innocence of a great dream.
Center for World Health, Department of Neurosurgery, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA
Correspondence Address: Jorge Lazareff Center for World Health, Department of Neurosurgery, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA
In the US the incidence of myelomeningocele (MMCL) is low, about 1/1200 live birth. This could be due to an active public health plant that mandates fortification of food with folic acid, and it also can be due to that in the US abortion is legal.
In my opinion, and I state it with out anything else but empirical observations, young couples are not fully aware that they need to take folic acid before becoming pregnant. Even though there is clear evidence pointing towards the advantage of folic acid on reducing the incidence of MMCL we should not fall into easy comfort. There may be other factors that need to be defined.
That abortion is legal has created a particular situation in the US, that of prenatal consultation. The parents alerted by the findings on the ultrasound schedule and appointment with a neurosurgeon sometimes together with a neonatologist and a neurologist. In the meeting the implications of a lesion sac filled with fluid in the bac of the fetus are discussed. I say lumbar or lumbar sacral area because we I can’t remember the last time I saw a child with a thoracic MMCL.
We all understand that the hurried meeting is called to help the family make a decision within the time frame when abortion is permitted. I conduct the conversation not as if I was reading from a textbook. My first words are to congratulate them for the child, then enquire if we now the gender of the unborn and even if it their first child and even if they have thought of a name. I don’t minimize the seriousness of the lesions, stress on the absolute certainty of sphincter damage, I am more reserved about the putative motor impairment but still I never give the impression that the child will not have to face some handicap. I also discuss hydrocephaly, I mention the complications but I also say that in our hospital we have a low, 2%, shunt infection rate.
In essence, while not driven by any religious believe I am more positive than dry objective. I a aware of a study conducted by Mc. Lone in Chicago were he states that very few parents repent from having allowed the child to be born.
In our hospital Dr. Bernard Churchill, a distinguished pediatric urologist who dedicated his skills to the well being of children with MMCL, advices cesarean section but we leave that decision to the obstetrician.
I don’t recall receiving a call from an hospital were the child was born about how to care the wound. Every child has always arrived with the wound gently covered with a soft pad and already with IV ATB.
I schedule the surgery as soon as possible without relining it. But if the surgery will be delayed 24 hours I act more forcefully, even if the child is well cared after at our state of the art neonatal unit.
I got this obsession from the Children's Hospital in Buenos Aires. We had this rule that if the surgery was delayed for more than 24 hours, even in the absence of signs of infection, we placed the child in a 2 weeks ATB treatment before repairing the defect.
Once the child is in the OR I don’t rush anybody and anesthesiologists and nurses dictate the pace. We take extreme precautions with the placode.
Certainly we don’t scrub it.
If the sack is intact I puncture it and aspirate the fluid, a lumbar puncture of sorts. I do this albeit I never retrieved infected fluid and even when I don’t wait for the results for starting the surgery.
I use the operating microscope for didactic reasons. I want the resident to be aware of every step of the procedure.
I incise the sac, aspirate the fluid through a cotton and proceed to dissect the placode. I handle it as it was viable tissue. I keep wet to reduce the noxious effect from the heat coming from the microscope lamp.
I find roots branching of the placode that seem to go nowhere. If they truly don’t go into the canal I divide them but with trepidation.
I also have doubts when facing a “secondary” placode. It looks as an independent island of the defective cord from where even some roots come out. I try to preserve both placodes, but if I see that both will be crammed in the dura sac I remove the smaller one. Examining patients after surgery I did not find added deficit to what was expected following the level of the lesion.
At hour six I always find a venous cluster that I divide.
I don’t suture the placode. I close dura, if the placode is wide for the canal I use a dura patch. Whereas when I was thought to close muscle, after my time in Cape Town with Warwick Peacok I switched and don’t close the muscle unless the dura plane is imperfectly closed.
Due to the nature of the patients we see here in the US I don’t have a strong opinion about early corpectomy. I know that Graciela Manucci favors it.
I close skin aiming at having the less possible tension. I don’t hesitate in restoring to linear or “Z” release incisions.
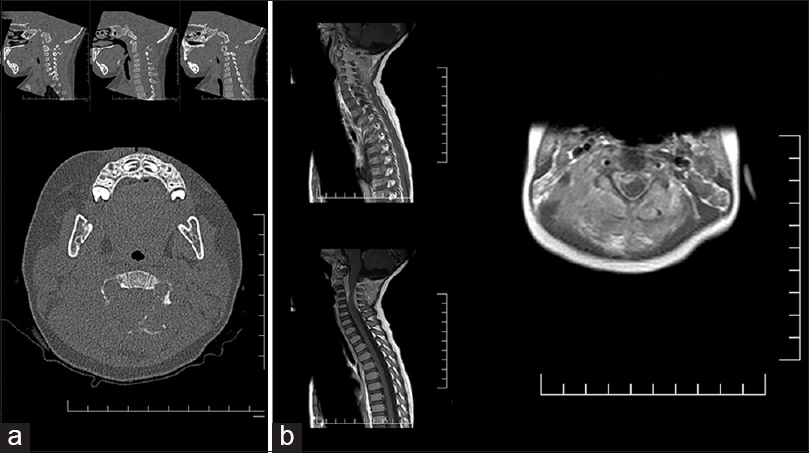
After surgery I don’t request a spine MRI. We place the child on ATB for 10 days after surgery. We monitor with ultrasound the condition of the lateral ventricles.
We shunt only after signs of intracranial pressure are present. But, I adhere to the principle that tension equals pressure times two the area. T = P × 2A. Thus I react to any enlarged ventricular surface and aim at reducing the tension on the brain by shunting. The intellectual development of the child is paramount in my book. I certainly have deep respect for my colleagues who wait for definitive signs of intracranial pressure.
Every child with MMCL has a tethered cord. Not everybody has symptoms. I pay attention to deterioration of motor and sphincter and above all to pain. I untether once, in the past I kept on untethering and I have to conclude that this can be futile. It goes without saying that each case deserves a through assessment independently of preconceived ideas. When I untethering I always use an artificial dura patch.
In the presence of symptoms I first revise the shunt. The foramen magnum is already enlarged so I don’t see the point in further enlarging it.
Of note, I have seen patients without Chairi II, thus I am not that convinced that it is a phenomenon related to fetal surgery.
The follow up is multidisciplinary. I pay special attention to academic performance. As stated above the intellectual strength is the best tool that the child has to face the world. A drop in academic performance can the only sign of a shunt malfunction or be secondary to tethered cord pain.
The attached video summarizes our technique. While I use dura patch I don’t have absolute evidence that it reduces the incidence of symptomatic tethered cord.
En Estados Unidos la incidencia de mielomeningoceles (MMC) es baja. Alrededor de un caso por cada 1200 nacidos vivos. Esto puede ser resultado de una activa prevención con ácido fólico, como también puede ser resultado de que el diagnóstico prenatal permite el aborto legal del feto. En mi opinión, y esto lo digo sin casi ninguna base de datos mas que los empíricos de observaciones entre la gente joven con la que he conversado ora como médico, ora como amigos de mis hijos, casi nadie está enterado de que deben de tomar ácido fólico durante la edad de concebir. Hay campañas de salubridad estatales, como la conducida en Texas, que ha disminuido la incidencia a través del suministro de ácido fólico. Lo que quiero decir es que tal vez existen una gran cantidad de factores que afectan la incidencia de la espina bífida abierta y que no debemos apoltronarnos en el confort de pensar que la solución del problema es un asunto de la futura madre cumpliendo su deber.
Que el aborto terapéutico sea permitido da origen a una situación particular; la entrevista prenatal. Los padres alertados por el obstetra de los resultados del ultrasonido de rutina tienen una reunión de consulta cuyo núcleo lo componen personal de trabajo social, neonatólogo y neurocirujano. Algunas veces se han sumado neurólogos. La intención de la entrevista es la de informar acerca de las posibles consecuencias de lo que el ultrasonido muestra como una bola de líquido a la altura de las vértebras lumbares. Acá debo de agregar que en general son lumbares bajas o directamente sacras, casi no vemos casos de lumbares altas y menos torácicas.
A nadie se le escapa, dada la premura de la reunión, que la información obtenida ayudará a la familia de decidirse a favor o en contra del aborto.
Ahora bien, la conversación con la familia nunca tiene el monótono discurso de quien lee un libro de texto. Nadie recita las incapacidades previstas de acuerdo al supuesto nivel de la lesión, casi siempre con gestos, el comentario. Me explico; Al recibir a los padres a veces con familiares, uno les da la bienvenida felicitándolos por la circunstancia del embarazo, le puede preguntar si saben ya el género del feto, si han pensado en nombres, si es el primerizo, si tiene hermanos. Luego uno pasa a enfatizar que los esfínteres estarán dañados sin lugar a dudas, y acá uno puede terminar la frase y con gesto severo enfatizar la gravedad de la condición o puede seguir y decir que los urólogos están trabajando continuamente para mejorar la situación de esos niños y que la función sexual de los varones está casi intacta y que las mujeres pueden tener hijos. Cuando se habla sobre la hidrocefalia uno puede enfatizar en las complicaciones o no, uno puede mencionar las infecciones y quedarse ahí o decir que en este hospital nuestro índice de infección no llega al 2%.
Personalmente, aún cuando no me anima ningún espíritu religioso, soy de enfatizar en lo positivo y me da alegría cuando meses mas tarde recibimos al niño en el hospital. Hay una estadística de Mc. Lone en Chicago (referencia) donde menciona que una minoría de padres se han lamentado no haber abortado.
He visto que mis colegas presentes en la reunión son más cautos, hasta pesimistas, esto tal vez para compensar mi optimismo. Demás está decir que soy consciente de esta postura mía y que más de una vez trato de enfatizar en que nadie esta juzgando una decisión y mi manera de hacerlo es situarme al mismo nivel que todos los médicos y trabajadores de salud presentes. Claro que, y acá está otra vuelta de tuerca, soy el único que habla español y eso de inmediato me da una ventaja sobre mis colegas, aún cuando todos hablemos inglés.
Finalmente llega el día. En nuestro hospital el Dr. Bernard Churchill, nuestro urólogo aboga por el parto por cesárea (la literatura y referencia acá no esta clara) pero yo lo dejo la decisión en la experiencia de los obstetras. Lo hago por respeto profesional, pero me arrepiento de no haber sido mas contundente en expresar que el paso de la médula desnuda por el canal vaginal agrega un componente negativo a una situación ya precaria.
No recuerdo haber recibido una llamada desde el hospital donde nació el niño sobre los cuidados de la herida. Siempre han llegado envueltos en gasa húmeda y con antibióticos endovenosos, generalmente vancomicina. El la unidad de cuidados neonatales hacen un exámen físico para eliminar malformaciones de otra naturaleza. El caso mas común es que la espína bífida y la hidrocefalia son las únicas. Si el niño ha nacido en nuestro hospital busco al padre y le informo sobre mis impresiones. A veces nos conocemos de la visita informativa de meses atrás. Programo la cirugía y si es posible hablo con la madre también. Meses mas tarde me confiesan que tienen un vago recuerdo de mi visita y que básicamente no entendieron nada de lo que dije.
Programo la cirugía para las próximas horas. No amenazo al quirófano con pasarla como urgencia, porque después de décadas ya saben que podemos llegar a un consenso para operar el niño en las primeras cuatro horas de vida. Si estuviera en un hospital de adultos no tendría inconveniente en ser mas enfático ya que una cirugía programada en un adulto la puedo “saltear” con mas facilidad. En mi hospital la anestesia será dada por un especialista en pediatría y será casi siempre realizada en un quirófano asignado a niños, por lo tanto a veces desplazaríamos a un niño para operar a otro, y eso trato de evitarlo. En esencia, el respetar la profesionalidad de nuestras enfermeras encargadas del quirófano hace que ellas mismas manejen la situación sin que nadie salga ostensiblemente preocupado.
Ahora bien, ¿de dónde me viene esa tara con que el cierre debe de hacerse lo mas pronto posible? Después de todo la herida ha estado abierta por horas y el daño supuestamente ya esta hecho desde hace rato. Lo atribuyo con certeza a mi entrenamiento en el Hospital de Niños Ricardo Gutierrez en Buenos Aires. No éramos maternidad, los niños nos llegaban del cono suburbano, la infección de la herida era una posibilidad y habían reglas bien estrictas acerca del margen de tiempo. No me las acuerdo ahora pero si el niño pasaba las 24 horas de nacido sin haber sido operado se programaba de segunda después de dos semanas de antibióticos.
Ni por asomo llegamos acá a estar cerca de ese margen, pero la impronta me ha quedado y niño que llega a mi hospital hoy se opera hoy.
Hay ocasiones en que los anestesistas tienen dificultades en conseguir vías de acceso. Hubo un niño que lo ingresamos al quirófano a la hora de haber nacido y lo empezamos a operar cuatro horas después.
Los anestesistas trabajan con el niño en decúbito lateral y si tienen que ponerlo de espaldas para la intubación lo hacen rápidamente y a veces uno de ellos levanta la espalda del niño para que no presione contra la camilla.
Cuando en decúbito prono mantengo la lámpara cialitica alejada del niño para disminuir la energía térmica.
Una vez descubierta la herida pongo una gota de solución fisiológica para mantenerla hidratada.
Preparamos el campo quirúrgico tratando a la médula expuesta como tejido viable. No aplicamos ningún desinfectante sobre la plaqueta medular, aún en casos cuando el niño tiene casi un día de nacido.
El límite del campo quirúrgico es al menos 10 centimetros de los bordes de la herida. Aún cuando no pienso prima fascie que necesitaré incisión de descarga preparo los campos de la misma manera. Si la incisión de descarga necesitará un trabajo mas complejo alerto a mis compañeros de cirugía plástica y ellos analizan la situación y determinan
Si el saco está intacto hago una breve punción “lumbar” del mismo para juntar líquido y determinar si hay ya una bacteria presente. Nunca he tenido un líquido con cultivo positivo. El análisis de las células y la glucosa de ese líquido es muy disperso como para sacar conclusiones.
Uso el microscopio quirúrgico con fines puramente didácticos. Como expresé en el primer párrafo no tenemos mas de cinco o seis casos por año y quiero que los residentes comprendan y comprehendan la patología.
Hiendo el bisturí exactamente al borde de la plaqueta.
Toda la succión del líquido es a través de algodón. La esencia es que la plaqueta y los raíces nerviosas son tejido viable.
Con tijera delicada completo dos medialunas, una a cada lado de la plaqueta, y luego diseco con extremo cuidado la parte cefálica del defecto. He visto casos en los que la plaqueta fue amputada del resto de la médula. Y acá cae de nuevo la pregunta, ¿afecta eso al niño?, en otras palabras, ¿la plaqueta es tejido fisiológicamente viable?. Yo pienso que si, de ahí los cuidados extremos en manipular el tejido como si fuera el lóbulo temporal izquierdo.
La parte que me ha preocupado mas de una vez es la de que hacer con esas raíces que salen de la plaqueta y parecen que van a ningún lado. Aquellas que penetran el plano de dura las corto, aquellas que entran al canal raquídeo las respeto. PERO, están aquellas que efectivamente van al canal raquídeo y que sin embargo se originan en una isla independiente de la plaqueta medular. Me explico, tenemos una plaqueta principal y otra periférica, casi como un satélite. De las dos salen raíces vigorosas y anatómicamente correctas. Acá temo apelotonar la plaqueta principal con la satélite y acabar estrangulando ambas. Entonces opto por eliminar la periférica, plaqueta y raíces. Nunca he tenido casos en que ambas plaquetas sean similares en tamaño o densidad de raíces.
Las trabéculas de aracnoides que puentean entre la plaqueta y el plano de dura las diseco.
Una vez que la plaqueta reposa en el canal raquídeo exploro visualmente el canal raquídeo superior al defecto. Estoy alerta también ante la posibilidad de un espolón de diastematomielia. Me ha pasado una vez y se me quedó la impronta.
Mantengo la plaqueta hidratada con solución fisiológica. Esto sobre todo por el calor emanando de la luz del microscopio.
Para el plano de duramadre hiendo el bisturí casi perpendicular al tejido, hasta sentir que estoy sobre la fascia del músculo. Con tijera completo el giro de disección.
Siempre hay una vena de drenaje a la hora seis, o en el centro del eje caudal. La coagulo, también coagulo casi todos los vasos que están interpuestos con la tarea de disección. No deja de preocuparme la isquemia del tejido nervioso y uso criterio. Por fortuna no hay vasos que directamente irriguen la plaqueta.
El cierre de dura es directo siempre y cuando la relación entre contenido y continente sea amplia. Si hace falta pongo un parche para permitir libertad en el canal medular, para no estrangular la plaqueta. No ha impedido la médula anclada en esos pacientes pero subjetivamente creo que su evolución ha sido acertada.
En el Hospital de Niños de Buenos Aires aprendí a cerrar el plano de músculo. Cuando estuve en el Red Cross War Memorial Children´s Hospital en Ciudad del Cabo vi que Warwick Peacock no lo hacía. Opté por la última. Se me hace mas noble con el tejido.
Sigue la maniobra de Valsalva que a veces puede ser reforzada por el anestesista ejerciendo simple presión sobre la fontanela anterior.
Para el plano de piel, aproximo con tres puntos los planos subcutáneos y luego reduzco el grosor del labio final de la herida. Todos queremos esa cicatriz linear sin repulge. Prefiero el repulge.
Cuando no hay piel redundante y tengo que cerrar piel sin incisión de descarga no me aflige mucho la palidez que noto allá donde la piel ha tenido que ser estirada hasta el máximo para llegar hasta el otro borde de la herida. Acá hay criterio quirúrgico que es difícil de transmitir al residente, pero en síntesis, un poco de ojo de cirujano, entidad metafísica desnuda de toda pretensión científica es útil.
Rara vez el diámetro de la lesión es tal que hay que pensar en incisiones de descarga. Mi experiencia me da solamente para una simple zetoplastía. Como comenté antes, el “ojo de buen cubero” nos da una idea de las dificultades del plano de piel.
Mi amiga Graciela Mannucci tiene gran experiencia en canales raquídeos complejos donde a veces hay que hacer corpectomía. Yo tengo escasa y nula. Esto tal véz porque la naturaleza del Mielomeningocele ha cambiado en EEUU. No vemos casos torácicos ni defectos muy grandes, ni ninguna de las otras variantes. Tal vez la dieta con o sin ácido fólico haya influido en la anatomía.
Continuamos con los ATB preoperatorios por una semana.
A veces los neonatólogos en su entusiasmo ordenan una resonancia magnética del neuroaxis. Yo no lo considero necesario. El niño tiene Chiari en la gran mayoría de los casos. La hidrocefalia se diagnostica por clínica y se confirma por ultrasonido.
Raros son los casos que no necesitan drenaje ventricular.
La hidrocefalia, yo adhiero a la idea de que T (tensión) = Presión × Area (2). O sea que si el área del ventrículo es grande aún cuando no crezca y no tenga presión intracerebral cínicamente expresada prefiero reducir el área del ventrículo. En otras palabras, estoy un tantito más inclinado a colocar que la válvula que a no colocarla.
Influye en esto que los pacientes tienen seguro médico y que nuestro de índice de infección es bajo.
Médula Anclada, en esencia todo paciente con MMC tendrá una médula anclada radiológicamente. Actúo sobre ella solamente cuando la evolución motora del niño evidencia un retroceso o cuando hay intenso dolor de espalda. Por un largo tiempo estaba a favor de la re-re, ya no mas. Si los problemas no se han solucionado con un desanclaje no se solucionarán con un segundo.
Para esta cirugía uso un parche y obsesivo monitoreo. Algunos casos son simples, un simple incidir sobre la cicatriz de la plaqueta despegándola del plano y de piel pero otros casos no son tan sencillos ni satisfactorios.
Chiari II Casi todos los pacientes tendrán Chiari II. Nota al margen, he tenido pacientes que no han desarrollado Chiari, lo menciono porque este es un justificativo para la cirugía fetal pero la ausencia de Chiari no es exclusiva de los casos intrafetales.
En casos de Chiari I, los no relacionados con MMCL, aumentamos el tamaño de la fosa posterior. En casos de Chiari II el agujero magno ya está agrandado, no tiene sentido en descomprimir. Por un tiempo era partidario de colocar un parche de duramadre. Ahora reviso la derivación ventricular. Si, aún cuando no hay ventrículos grandes considero que el desequilibrio fisiológico de una derivación que no funciona óptimamente precipita los síntomas.
El paciente con Mielomeningocele es un niño que tiene una discapacidad motora y sensitiva de los miembros inferiores y de los esfínteres. Su principal tesoro es la su condición de humano inteligente y pensante y creador. Incluyo en el seguimiento clásico la actividad académica. Le pido a los padres que estén alertas ante cualquier disminución de las notas en la escuela. En tal caso considero revisar la válvula, pero sobre todo enfatiza en el niño o adulto joven y en los padres que la capacidad intelectual no ha sido disminuida.
En el video se demuestra la técnica que usamos. Acerca del parche de dura, no tengo evidencia conclusiva que reduce la incidencia de médula anclada sintomática.
The Hospital Infantil Manuel de Jesus Rivera, La Mascota is the main pediatric referral hospital in Nicaragua. Since 2010 there is a division of Neurosurgery within the Department of Surgery. Two Neurosurgeons staff the division.
We see an average of 44 new cases of neural tube defects (NTD) per year. I have nothing but praise and gratitude for the neonatology department at La Mascota. Every month we perform 3 or 4 four new cases of myelomeningocele (MMCL). The great majority are children of very young women; some are 15 or 16 years old. They come from distant rural areas but many are from the capital as well. Managua is the most densely populated region of the country.
In Nicaragua there is no national program for fortifying flour with Folic Acid. What we have in place is a program for providing folic acid to pregnant women when it is known that the malformation has already occurred. I say this acknowledging as well that there may be other factors besides folic acid deficiency responsible for NTD. In any case the importance of folic acid in preventing NTD is such that our government has to implement fortification of flour with folic acid.
There are many differences between high-income countries and low and middle -income countries about how to care for children with NTD. Along the years some things have remained the same but some have changed for good. When I trained an attending in Masaya told me that children with NTD had a very short life span, implying that we should not waist resources on them.
In our country there are barriers to care. We have only two Neurosurgical centers in the country. Many children and their mothers have to travel for many hours in deficient conditions. When the child arrives, many days after birth, a yellow patina of reactive tissue covers the placode. We need to sample it and wait at least 3 days to determine if it is infected. If it is infected, we request the help of the department of Infectious Diseases and the child is for an added 3 weeks in house. We have spread the word among our colleagues from rural areas that it is imperative that the newborn with NTD be referred to us as soon as possible. Still this is not possible in children coming from the Atlantic coast.
In children with infected placode we perform a ventricular tap. Sometimes we obtain dense pus. This year I had three cases of a child with severe ventriculitis but without any signs of infection. The babies were being breast-fed without evidencing any sign of distress. This has leaded me to ponder if the CSF has a role in the immune system or that the ependyma is a strong barrier that prevents the spreading of infection.
The patient is usually received with the defect covered by wet gauze directly over the placode and dry gauze on top. The child is then admitted to the neonatal ward.
A multidisciplinary team of pediatricians, surgeons and orthopedist then sees the child for detecting associated anomalies. We perform abdominal ultrasound and head ultrasound, echocardiography and routine laboratory testing including electrolytes.
The neonatologists always prescribe Vancomycin and Ceftriaxone. Surgery is scheduled for the next day, first case. This of course provided that the child is not infected. The anesthesiologist evaluates the child the night before surgery and directs 4 hours NPO.
The afternoon before surgery I visit the mother. I am accompanied by a pediatric surgery resident. We don’t have Neurosurgery residents. I explain to the mother in the most warm and direct fashion the objectives of the surgery, the complications, the long-term prognosis of the child, the certainty of sphincter dysfunction and I raise the possibility of the child needing a shunt.
It has always impressed me how happy the mothers are by the simple fact that the cosmetic aspects of the pathology will be corrected, I see this when they come back to clinic. This has inspired me to do the best that I can for the child.
In the operating room I turn off the air conditioning. We don’t have thermic blankets and I don’t want to risk hypothermia.
General anesthesia, prone, over two rolls under the shoulders and hips. I personally sterilize the area. Furthermore, I requested that the day before the back of the child be washed twice with a 4 hours interval with a solution of hibiscrub, saline and alcohol. Certainly I emphasize that the placode not be included.
I prepare the surgical area around the placode with soap and alcohol. I scrub gently to do avoid damaging the sensitive skin of a baby. I do this twice with patience, lots of patience. Then I cover the field with sterile drapes and then I scrub. I wear double gloves, I may not be aware if one of them has a puncture. I carefully with wet gauze remove powder from the gloves.
With sterile gauze held with a forceps I proceed to swap with betadine the skin up to 4 centimeters away from the edges of the defect. I wait until it dries, and finally I stich the drapes to the skin. Our surgical drapes are reusable. I don’t use mono-polar coagulation or suction.
I am a Christian, not a good one but a believer. Briefly and in silence I entrust the surgery to God. Then with the permission of the anesthesiologist I start with the bipolar cauterizing, less than 1 centimeter of tissue between the placode and the epithelium. I start at “9’ o clock”. I choose this location to start the surgery because the risk of hitting a rootlet is minimal and because I am right handed.
I take this opportunity to teach the surgical resident who is assisting me the difference between the three tissues, placode, epithelial membrane and skin. One day this knowledge may be useful for some child.
I incise with skin blade #15 over the cauterized area. In large lesions the CSF bursts out. I patiently soak the fluid with gauze. Then with a Kelly forceps I widen the opening and explore the inside of the lesion to identify the stem of the cord, the dorsal roots and the blood vessels.
Slowly I proceed to dissect the placode from the surrounding tissue. I proceed cephalically and caudally but up to the midline. After completing the lateral dissection I focus on the cephalic portion with extreme care because it is there that the cord dives into the canal. Caudally I try to preserve a cluster of veins that can always be found precisely were the placode attaches to the skin. Considering the child's age the bleeding can be significant.
Once the placode is free on the bed of the defect I proceed to restore its cylinder shape by approaching its edges with a 5-0 Prolene noncutting needle. I try to shape the placode in such fashion that it will be loose in the bed of the defect. I constantly irrigate the surgical field with saline solution.
The next step is to identify the dura mater. I dissect the dura aware that I can here is where the risks of blood loss are higher. This is because the underlying fascia and muscle are well vascularized. So as I advance with Kelly and scissors I carefully cauterize the bleeding vessels. I do this trying not to damage the vascularization of the muscle and skin layers that will be crucial for the healing of the wound. Also at hour 6 is where there is a condensation of blood vessels. I dissect portion of dura from both sides before crossing the midline. I then proceed to dissect the bottom of the dura. This is a simple step. The fat layer about the bottom of the dura sac facilitates the dissection.
The closure of duramater is a crucial step of the surgery. I make sure that the dura is not under tension after I sutured it with Prolene 5-0 in a noncutting needle. I request the anesthesiologist to perform 3 Valsalva maneuvers and we all carefully check for CSF fistula.
I do not close fascia. I used to do it in the past. I admit that it adds a protective layer to the dura and the placode, but I have seen wound necrosis and dehiscence in case where the muscle was flapped over the dura plane.
With a non-tooth Kelly forceps I handle the skin edges. Gently with my finger I dissect the subcutaneous tissue to loosen up the epidermis. If I encounter a blood vessel I dissect it as if I was doing a by-pass.
I keep the skin and the subcutaneous tissue wet with saline solution. Then I anchor each side of the wound with a stitch through the subcutaneous layer with a Vycril 2-0. This helps me define the axis of the closure and if it is necessary to loosen more the epidermal layer.
I favor closing skin vertically. If not possible horizontally or italic S, and if needed a “Z” shaped release incision.
Once I defined the axis of the closure I release the Vycril 2-0 stitch. I proceed to trim the devitalized edges of the skin. I reach a level at which the skin edges start bleeding. My assistant exerts gentle pressure on the skin edges while I rapidly suture the skin with Nylon 5-0 starting the process at. 5 centimeters from the edges of the wound. To achieve perfectly opposing skin edges I may thread a temporary Mayo or a Zarnoff. Unfortunately I may be wasting material.
After stitching the skin I press along the incision with rolled gauze to drain out pockets of fluid and blood.
I cover the wound with Tegaderm, which is transparent and will allow us to detect hemorrhage.
For me the objective of surgery has always been to achieve a hermetic closure of the wound.
I try to preserve the integrity of the neural tissue in extremis. But I also fear infection, and the consequences can be devastating. Thus I am less cavalier with the placode of those children who arrived weeks after surgery and the placode is already poorly vascularized if not plainly infected.
I keep in mind what my wise friend and teacher Jorge Lazareff told me once; we treat patients and not diseases.
There are no textbook cases. So while I am moved by the happiness of the mother when she anticipates the bulky mass will be removed I sacrifice a cosmetically skin closure by making release incisions that reduce the tension of the wound and secure and adequate wound healing.
At the end of the surgery, I explain the mother that the child needs to be prone or at least lateral to reduce pressure over the surgical area. I advice her that when breast-feeding she holds the baby with a makeshift doughnut over the surgical area.
If possible we don’t remove the OR wound dressing for at least 48 hours. If after the first dress change I see that the wound is clean and healthy I just cover it with sterile gauze. The neonatologists manage the ATB regiment.
El Hospital Infantil de Nicaragua Manuel de Jesús Rivera, La Mascota, es el principal centro de referencia pediátrico a nivel nacional, donde desde hace 3 años cuenta con la división de Neurocirugía Pediátrica, adscrita al servicio de cirugía pediátrica, laboran ahí 2 Neurocirujanos.
Se han recopilado las estadísticas de los últimos 3 años en lo relativo a los defectos del cierre del tubo neural (DCTN), las cuales demuestran que se reciben en promedio 44 nuevos casos por año, en este particular agradezco al servicio de neonatología de este centro.
se realizan por mes 3 o 4 nuevas cirugías para el cierre de mielomeningoceles, nuestros pacientes son hijos de madres muy jóvenes, la inmensa mayoría menores de 20 años y un buen porcentaje son adolescentes de 15 o 16 años, provienen de los departamentos más alejados de la capital, sin embargo la capital aporta un buen porcentaje de pacientes por condensar la mayor cantidad poblacional del país
En Nicaragua no existe un programa sostenible de fortificación con ácido fólico de los alimentos de mayor consumo, esta normado aportar ácido fólico durante el embarazo, algo que en lo personal considero firmemente debe de cambiar, por razones embriológicas y fisiopatológicas que demuestran que los defectos del cierre del tubo neural ocurren en etapas muy temprana de la embriogénesis, quizás cuando la madre aún no está segura de su embarazo, y de algo estoy seguro que la mayoría de los embarazos en el país son no planificados. Todo lo anterior alimenta la persistencia de la aparición de esta patología, seguro estoy también hay otros factores satélites que contribuyen en mayor o menor medida a esta prevalencia.
Creo tenemos grandes diferencias en cuanto al manejo de los pacientes con defectos del cierre del tubo neural, en relación a países Latinoamericanos y por supuesto en relación a países con grandes ingresos económicos donde creo este ya no es un problema tan frecuente por la implementación del ácido fólico en los alimentos, diferencias que se estrechan quizás en relación a países de la región centro americana, sin embargo hemos cambiado el tabú de que los niños con mielomeningocele por ejemplo, tenían mal pronóstico para la vida y mal pronóstico funcional, eso lo recuerdo muy bien, pues en los tiempos de mi internado, en mi ciudad Masaya, cuando teníamos un paciente con esta patología mi médico de base lo transfería con la mayor premura e indiferencia y recuerdo su explicación: ese bebe va a morir pronto.
No obstante hay grandes barreras que nos golpean con dureza: las infecciones, esto ocurre principalmente por el transito que tiene que vivir el binomio madre hijo, ya que solo contamos con 2 centros que tienen Neurocirugía y al nacer en otro hospital debe ser trasladado lo cual tarda en ocasiones y en dependencia del lugar de origen, varios días, al recibirlo la placoda está ya con una capa amarilla, con material fibrinoide, esto nos obliga a tomar una muestra de este material y esperar al menos 3 días para obtener un resultado, después de ello tenemos 2 caminos: si el cultivo es negativo procedemos con la cirugía, si el cultivo es positivo solicitamos el apoyo de infectologia y eso nos hace esperar al menos 3 semanas, por tanto hemos promulgado en todo el país la referencia ultra temprana, pero ha sido imposible en algunos casos por lo lejos del origen del paciente como es el caso de la zona del atlántico.
Si el paciente tiene una infección en el sitio del mielomeningocele, y recibe tratamiento correspondiente, al finalizar este realizamos un nuevo cultivo para definir la cirugía, en el mejor de los casos es negativo y lo intervenimos a la brevedad.
A todo paciente que es recibido con más de 3 días de vida o tiene datos francos de infección de la placoda, o está roto el mielomeningocele, los pediatras han optado por realizarle punción ventricular transfontanelar, esto en algunos casos ha demostrado infecciones ventriculares graves, obteniéndose liquido purulento, estando el paciente asintomático, es decir sin fiebre, tolerando bien el seno materno, con buen estado general, sin datos clínicos de sepsis, y al realizar la punción se ha obtenido pus. Esto me ha llamado fuertemente la atención, pues en este año he tenido 3 casos, con un asombro que me es difícil escribir, he obtenido pus densa y el paciente aparentemente está bien, lo cual genero una pregunta en mi mente acerca de la función del LCR: es este un medio con propiedades inmunológicas que hace que la infección se delimite al ventrículo, o el revestimiento ependimario ventricular es una barrera suficiente para no diseminar la infección.
El paciente por lo regular es recibido con una cubierta en la zona del defecto con gasas húmedas y encima una gasa seca, se ingresa a sala de neonatología, se le realizan rutinariamente, ultrasonido abdominal, ultrasonido transfontanelar, radiografías de la columna con énfasis en la zona del defecto, ecocardiograma, exámenes en sangre que incluyen biometría hemática, tiempos de coagulación, plaquetas, tipo y RH, química sanguínea, es valorado por ortopedia en el caso de pies equino u otra malformación que lo amerite, así como valoración por cirugía pediátrica en el caso de malformaciones anorrectales que las vemos con regular frecuencia.
En la totalidad de los casos Neonatología opta por usar antibióticos a su ingreso: ceftriaxone y vancomicina. Al ser evaluado por neurocirugía y no haber una contraindicación para la cirugía, esta se programa para el día siguiente en primer turno con un ayuno de 4 horas, la noche anterior es visitado por el anestesiólogo.
La tarde antes de la cirugía converso con la mama, la llamo a parte, llevo a mi residente, que es de cirugía pediátrica pues no hay residencia de Neurocirugía en este centro, y hablamos a cerca de los riesgos y los beneficios de la cirugía, la patología, el pronóstico y que puede pasar durante y después del procedimiento, hablamos de la posibilidad de que el niño desarrolle hidrocefalia, al final las mamas aceptan de buen agrado la cirugía pero sobretodo con gran esperanza, pues se bien que ellas no han comprendido gran parte de la información que les he dado a pesar de explicarles en un lenguaje supremamente sencillo y con muchas analogías, pero su esperanza está centrada en no verles el defecto, quizás no tanto en el pronóstico funcional y el hecho de que probablemente tendrá trastornos urinarios y defecatorios asociados, sino más bien en la parte cosmética, pero es un aliento que me estimula a hacer lo mejor que puedo, esto lo veo después en la consulta externa, las madres se ven felices cual niño sin defecto
En el salón de operaciones usualmente apagan el aire acondicionado por no tener manta térmica y el riesgo de la hipotermia, se verifica la vía venosa, los datos del paciente, la nota de consentimiento informado, bajo anestesia general, en prono, con dos rollos de tela bajo el torax, del grosor de los brazos del paciente, procedo a lavar el área quirúrgica, antes lo hacían las enfermeras del salón de operaciones, en mi país les llamamos técnicas quirúrgicas y las circulares, pero de un tiempo acá prefiero hacerlo yo pues creo es muy importante esta parte de la cirugía ya que muchas de las infecciones post operatorias están relacionadas con gérmenes de la piel, y como las infecciones son muy frecuentes, he normado también que todo paciente que yo vaya a intervenir debe recibir una limpieza del área quirúrgica dos veces el día previo a la cirugía, con 4 horas de intervalo cada limpieza, esta debe realizarse con jabón hibiscrub, alcohol y solución salina, por supuesto sin utilizar ninguna solución iodada en el área del mielomeningocele es decir se lava la piel únicamente.
En sala de operaciones lavo con jabón, alcohol, luego seco y repito el procedimiento, en total dos veces sin hacer fricción y no lastimar la piel o causar eritema, se realiza con gentileza y paciencia, una vez lavado cubro con un campo estéril, y voy a lavar mis manos, usamos dobles guantes en toda cirugía teniendo cuidado de retirar el polvo de los guantes con una gasa húmeda, lo de lo dobles guantes lo he implementado a partir de que es fácil de que se rompan los guantes y no me doy cuenta por estar concentrado en el procedimiento quirúrgico, una vez me ocurrió y quien se percato fue el asistente quirúrgico.
Al iniciar, con una gasa estéril montada en una pinza, 1 cm alrededor y por fuera del mielomeningocele cubro con solución iodada (betadine), espero se seque, pues no tenemos ioban, 4 cms cuadrados alrededor del mielomeningocele, coloco campos estériles y los fijo a la piel con sutura una en medio de cada campo, esto evita que se deslicen los campos y se exponga más allá del área estéril, cubrimos con sabana hendida, desafortunadamente todos los campos y sabanas son reesterelizables, conectamos bipolar, no utilizo succión ni cauterio monopolar, este último en esta cirugía no lo encuentro tan necesario excepto en los casos en los que deba de realizar una cifectomia.
Soy Cristiano, no de los buenos, pero creyente, siempre en silencio y en brevedad encomiendo lo que hare a DIOS, luego con el permiso del anestesiólogo inicio colocando el bipolar sobre el límite del tejido epitelial y la placoda, esto lo hago siempre a las 9 del reloj, por 2 razones, una es que habitualmente es donde menos raíces nerviosas encuentro y dos porque soy diestro y me es más comodo. Me esmero para que el residente reconozca la diferencia entre los tres tejidos superficiales del defecto: placoda, membrana epitelial, piel displasica.
Sobre esta zona Coagulada, en menos de 1 cm, incido con un bisturí número 15, el LCR sale abruptamente más aun cuando los defectos son muy grandes, por lo cual minimizo esta apertura y absorbo este líquido con gasas, otra vez mucha paciencia, una vez que el defecto ha perdido su volumen, con una pinza Kelly amplio la apertura para observar dentro del defecto e identificar el cordón medular, las raíces y los vasos tantos periféricos así como los nutricios principales.
Despacio y con gentileza, con la pinza Kelly amplio poco a poco la apertura y corto la membrana epitelial, hasta donde me permitan las estructuras, si es un vaso periférico lo coagulo, si es una raíz trato de preservarla y de disecarla empujándola hacia la línea media para continuar bordeando el defecto y llegar a la línea espinal, a las 12 del reloj, en sentido cefálico. Igual procedimiento realizo del otro lado hasta llegar otra vez a la línea media, a las 6 del reloj, en sentido caudal, en esta zona siempre me detengo pues me encuentro con un nicho venoso importante, trato de no romperlo, pues casi siempre el sangrado es importante más aun considerando la edad del paciente. Una vez completado el corte del tejido epitelial en 360 grados alrededor de la placoda, esta ha quedado libre, procedo a irrigar con solución salina tibia, en el caso de que la placoda este sin datos de infección previa la invagino hacia la línea media formando un tubo, para ello utilizo hilo prolene 5.0 sin filo con sutura continua, luego siguiendo el cordón medular unido a la placoda, en sentido cefálico identifico el canal espinal, en este trato de posicionar el tubo reconstruido tratando de que quede holgado o al menos en línea media abocado a este canal, vuelvo a irrigar, haciendo énfasis en la hemostasia con coagulación bipolar.
Mi siguiente paso es identificar el tejido dural, el cual lo logro ver en las paredes disrraficas de las vértebras como un tejido perlado, denso o más grueso que la membrana epitelial supra yacente, identifico este límite, e incido con un nuevo bisturí número 15. La disección del tejido dural es siempre muy sangrante, quizás la parte más sangrante del procedimiento por la adherencia a tejidos mejor irrigados como los músculos, la fascia y la vértebra, por lo cual al ir disecando con la pinza Kelly también voy coagulando, teniendo cuidado de hacerlo del lado del tejido dural para no amputar vasos nutricios que serán importante para la cicatrización de la herida, el corte de este tejido dural lo realizo con un tijera muy fina, tijera de iris, que me permite también disecar al mismo tiempo. De igual manera completando los 360 grados con la idea clara que en la línea media a las 6 del reloj y en esta disección, es seguro habrán mas vasos sanguíneos y la probabilidad del sangrado es mayor por esta razon en este punto lo diseco bilateralmente al mismo tiempo hasta lograr desprender el tejido que quiero minimizando el sangrado, no siempre se logra pero el punto importante es tener en mente las zonas de sangrado. Una vez completado todo el corte, de toda la circunferencia, desprendo el tejido dural en profundidad para formar un bolsón, generalmente es un paso más fácil porque debajo hay grasa que permite el desprendimiento, es cuestión de utilizar una tijera fina que corte las adherencias visibles y casi siempre en el resto de la profundidad el tejido dural se diseca por sí solo.
Me aseguro que el tejido disecado que servirá como cubierta dural quede sin tensión, por lo cual diseco con más énfasis en la línea media donde se adhiere más firmemente y puede romperse, disecado ya, lo suturo uniéndolo en la línea media con prolene 5.0 sin filo, sutura continua, al terminar le pido al anestesiólogo realice maniobras de valsalva al menos 3 veces y le pido a mi equipo observen cualquier dato de fistula, en caso de no haber, irrigo y me detengo, observo la fascia, y el musculo y busco los puntos de mejor vascularización para protegerlos.?En lo personal no cierro el musculo, porque es una zona vascularizada que aporta a la cicatrización, y aunque el cierre de la capa muscular me da una cubierta más, he visto con mucha frecuencia la necrosis y dehiscencia de la herida asociada a este cierre, por esta razón hace varios años ya deje de hacerlo y con plena seguridad puedo decir que he obtenido mejores resultados, por lo cual el cierre de la capa dural creo es el punto medular en esta cirugía.
Luego con una disección sin dientes, tomo el borde la piel, la cual ha quedado libre, y también con gentileza y con la punta de una tijera metzembaum separo el tejido celular subcutáneo de la fascia muscular, esto lo hago en la línea media en sentido cefálico, profundizando en sentido paralelo al eje espinal, unos 4 cms, luego digitalmente completo esta separación y disección en 360 grados poco a poco, de tal forma que me detenga en las zonas de sangrados, la intención de hacerlo digitalmente es evitar cortar vasos nutricios que serán claves para la cicatrización, al encontrarme con un vaso, hago lo máximo por no cortarlo, lo diseco cual si fuese a realizar un bypass, si es posible o su calibre es grueso, continuo hasta liberar por completo la piel con el tejido celular subcutáneo en conjunto para preservar los vasos que por ahí entran a la superficie dérmica.
En dependencia de del tamaño, tipo y orientación del defecto, cerrare la piel. Puedo decir que más de la mitad de los casos logro cerrarlo verticalmente siguiendo el eje espinal, en el resto de los casos realizo un cierre horizontal, una Z plastia o un cierre haciendo una S hitalica. Antes de iniciar el corte de la piel irrigo con solución salina, mantengo húmeda la piel y el tejido celular subcutáneo sin derramar mucha solución salina sobre el bebe, y doy un punto de anclaje – referencia, subcutáneo acercando la piel, con vicryl 2,0. este me sirve para definir la orientación del cierre y para conocer mas o menos el grado de tensión que habrá al cierre, lo cual define si debo ampliar más la disección subcutánea hacia los costados, esforzándome en el hecho de que la piel se junte sin presión, o la mínima posible, esto depende definitivamente del tamaño del defecto, pues en los defectos pequeños la disección es mínima y todos los tejidos sufren menos, una vez que defino el eje del cierre y asegurándome que no hay tensión, corto el punto de anclaje – referencia y corto los bordes de la piel, más o menos 1 cm de grosor, ya que estos bordes son mal vascularizado, displasicos y es casi seguro de no hacer esto la cicatrización será mala y habrá una dehiscencia, veo que los bordes estén sangrantes, mi ayudante hace hemostasia al presionar estos bordes, mismo tiempo, con la mayor rapidez posible comienzo el cierre de la piel, esforzándome en que los bordes queden bien afrontados, con nylon 5.0 a medio cm de cada borde, doy puntos de mayo, separados, a veces zarnoff, intercalados con el objetivo de que los bordes queden afrontados y sin tensión, desafortunadamente tengo que utilizar sutura que luego tengo que retirar. Al completar el cierre, hago un poco de presión con una gasa como rodo en sentido cefalo caudal para drenar restos hemáticos.
El objetivo en esta cirugía siempre ha sido para mí el cierre hermético, sin embargo y a pesar de las controversias que puedan haber en la literatura, trato de preservar todo el tejido nervioso que pueda, no siempre lo logro, por ejemplo, a veces la placoda ha estado mucho tiempo expuesta, es decir la cirugía se realiza semanas después del nacimiento, y prefiero no correr el riesgo de una infección o reinfección, creo que lo que cambia definitivamente el pronostico funcional en estos niños o lo que lo define es que ocurra o no una neuroinfeccion y eso lo tengo siempre muy en cuenta tomando como premisa el adagio que un hombre sabio me dijo una vez: tratamos pacientes no enfermedades, me lo dijo mi estimado amigo y profesor Jorge Lazareff MD, esto porque no nos podemos apegar a texto todas las veces y siempre, siempre hay bemoles en la sinfonía de toda cirugía, a veces corto los extremos de piel sana para que el cierre quede sin tensión, la herida será más grande y mucho menos cosmética pero me aseguro que cicatrice bien
Al finalizar la cirugía, le explico a la mama que el niño debe de estar boca abajo o lateral y que en todo momento evite la presión sobre la zona quirúrgica, al amamantarlo que utilice una dona sobre la zona quirúrgica, le pido a los residentes no descubran la herida a menos que haya datos de sangrado, cubro la herida la mayor parte del tiempo con tegaderm, como es transparente me permite ver la herida sin descubrirla, usualmente la descubro a las 48 horas, el niño continua con los antibióticos ya prescritos por neonatología y ellos deciden el tiempo de dicho esquema, al curar la herida, si está completamente limpia solo coloco una gasa estéril y la cubro de nuevo, sino limpio con solución salina.
How to cite this article: Manucci G, Quednow Ev. Como Lo Hago Yo: Anomalías del Tubo Neural en Guatemala — Mielomeningocele Unidad de Espina Bífida e Hidrocefalia. Surg Neurol Int 10-Mar-2014;5:
In Guatemala the prevalence of Neural Tube Defects (NTD) is approximately of 2.34 per 1,000 live births. It represents the most frequent congenital anomaly. Surpassing cleft palate and abdominal wall malformation.
Considering that the country has an annual population growth rate of 2.8% we can infer that of 336,000 new born per year, 786 will have a NTD. The higher prevalence is in the NW region, perhaps related to the indigenous population and poor nutritional status.
Folic acid deficiency, mycotoxins such as fumonisin or genetic alteration of the metabolism of tetra hidro pholate reductase, key enzyme for folic acid metabolism, could be relevant.
The NTD are
Anencephaly
Spina Bifida Aperta (myelomeningocele) and Spina Bifida “closed”
Encephaloceles, anterior and posterior.
The most frequent is the myelomeningocele. While folic acid deficiency is the most common etiological factor it is believed that the high incidence that we have in Guatemala could be secondary to a genetic cause. We have seen many cases in the same family including a 30% of familiar cases where the individuals do not leave in the same region.
Since I arrived from my country Argentina to work in Guatemala I devoted my efforts to build a ward at the San Juan de Dios Hospital devoted to the care of NTD and Hydrocephalus and associated conditions. It is the only specialized ward in Central America.
In the unit we perform more than 250 surgeries every year, of which 65 to 70 are closing of myelomeningocele.
In Guatemala the barriers to care of children with NTD are social, economical and cultural. It is believed by the community and by health care workers that the children with NTD are crippled mentally retarded.
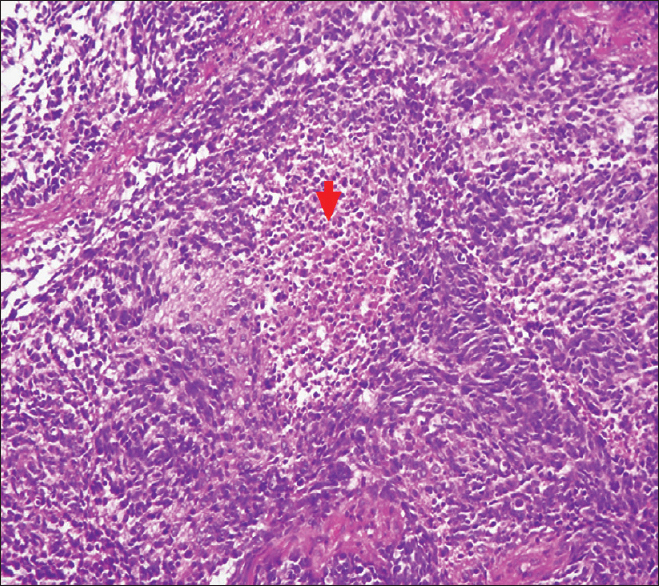
Is the most frequent and severe form of spina bifida. It is a complex anomaly that affects the central nervous system but also the renal and urinary system as well as the spinal column and the lower limbs.
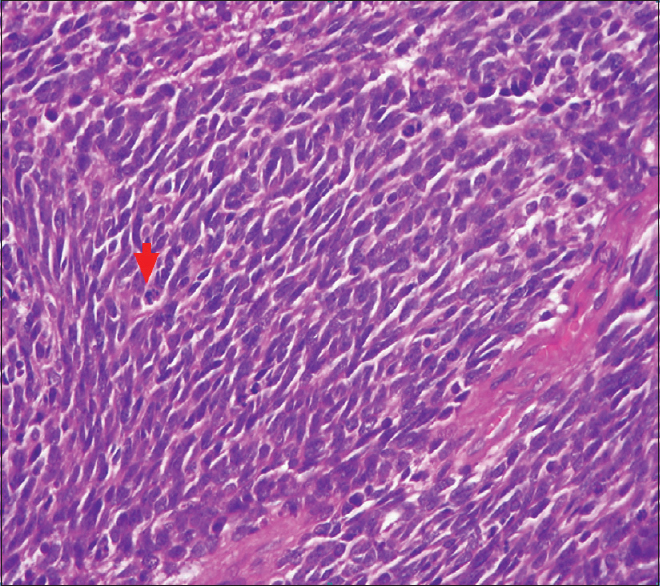
It has a central portion, the placode, which is pink or red, surrounded by a thin and transparent arachnoid and at the external margin by a transitional tissue, a very thin epidermis that is poorly vascularized and often inadequate for stitching.
Factors that affect prognosis
Prenatal diagnosis
In our country only 40% of pregnant women have access to prenatal ultrasound.
Considering that surgery within 12 hours of birth is crucial we have created a system through which the mother-often very young and not infrequently single and without family support-receives psychological support and orientation. This is not only for the immediate benefit of the mother but also for developing the welcoming of the child.
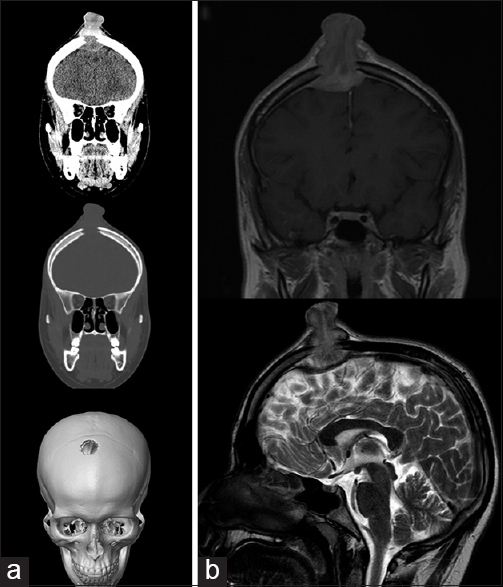
A geneticist determines if the lesion is an isolated incidence and a treatment plan is developed according to some genetic findings. Comes to mind the Jarcho-Levin Syndrome in which there is a malformation of the axial skeleton that compromises the respiration [ Figure 1 ].
Figura 1
Jarcho-Levin syndrome
Prenatal diagnosis allows us to plan for the surgery. At our hospital we have OR access limited to certain days. We strongly recommend delivery through cesarean section. If the myelomeningocele sac ruptures during delivery it increases the risk of sepsis and becomes an absolute emergency. In hospitals were there in no Neurosurgeon we recommend simple suture of the skin to reduce the exposure and the CSF fistula and to transfer the patient immediately to our unit.
Closure within 12 hours of birth
This to (a) avoid infection, particularly the dreaded Gram-negative bacteria. In our country children born with NTD are admitted in the regular neonatal unit where the criteria of sterile wound care is not perfectly implemented. Children born away from Guatemala City are transferred by land, often a 9 hours drive and they arrive dehydrated, hypothermic and in acidosis.
Follow up by a multidisciplinary team
This is so important that I consider it as relevant as surgery. After discharge the child is regularly followed by a team of Neurosurgeons, Urologists, and Orthopedic Surgeons specialized in spinal column, Pediatrician, Physical and Occupational Therapist, Social Worker, Pediatric Surgeon and psychologists specialized in Early Development. Appointments are tailored according to the needs of each patient.
We look for
Associated Anomalies
Hydrocephalus
Symptomatic Chiari type II
Secondary Anomalies
Neurogenic bladder
Urinary reflux
UTI
Incontinence
Skeletal deformities
Limb deformities
Hip luxation
Kyphosis and Scoliosis
Tethered cord.
Psychological support for the parents
The birth of a malformed child affects the family dynamics, it may generate rejection of the child and not infrequently we see mothers who abandon their child.
Continuous physical therapy
Two or three times a week. Starting as soon as possible.
With surgical knife and completed with Metzembaum scissors I incise at the boundary between the placode and the transition epithelium.
Detach arachnoid from placode
I perform hemostasis with “mosquito” with extreme care so not to impair epithelial irrigation.
If the placode is small and fits nicely in the canal I do not reduce its size. If the placode is large and wide I proceed to slice off the surface, the tissue that was most exposed to the environment. I utilize bipolar coagulation. Once I established that the placode is lying free of adhesion in the canal I proceed to close duramater.
For closing duramater I dissect the tissue with surgical knife and scissors. Closure with 4-0 Prolene. I request a Valsalva maneuver to confirm watertight closure.
I do not dissect or suture the muscle layer
I do dissect the subcutaneous layer up to 4 centimeters away from the edges of the wound. I bring the edges together and anchor them with few Vycril 3-0 stitches. Enough for holding the skin and then proceed to trim the redundant tissue. Finally I suture the edges with Nylon 4-0.
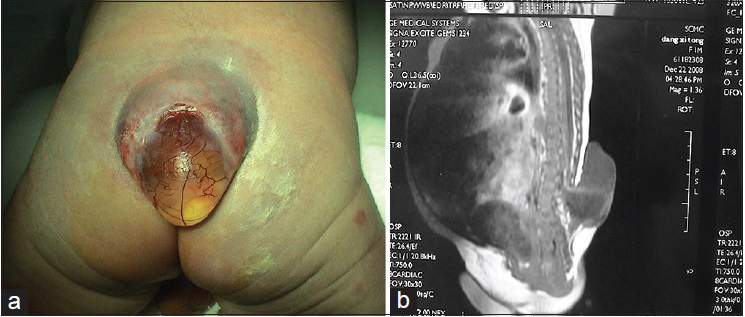
There is a large (30%) subgroup of patients in which the kyphosis is severe and the closure of the skin is difficult [Figures 2 – 4 ].
Figura 2
Grade 2
Figura 3
Grade 3
Figura 4
Dehiscence of surgical wound in a patient with kyphosis
Wide base. In this case I perform lateral release incision and if needed I flap the dorsalis over the dura sac.
Before surgery
Grade 2
The child has angulated Kyphosis. After closing the duramater I perform corpectomy. After corpectomy I approach with 2-0 silk the soft tissue that was about the vertebral body that was removed. In this fashion I reduce the angle of the defect and are allowed to close skin without tension.
Grade 3
It is the most difficult. It is a combination of Grades 1 and 2. It demands adequate perioperative support. For that reason I defer the surgery until the child clinical condition is stable factoring the risk of infection if the surgery is delayed.
Patients in which the kyphosis was not corrected and there is dehiscence of the surgical wound.
The progressive kyphosis decreases pulmonary capacity. It does not allow the child to sit comfortably and causes pressure wounds.
Present in 80% of the cases, although is only evident at birth in 20% of children.
I shunt only after evidence of progressive hydrocephalus. I agree with Dr. Maurice Choux on that in 30% of children Hydrocephalus associated with myelomeningocele detains its progression thus not requiring shunting.
I don’t place the shunt at the same time of closure of the myelomeningocele. It increases the risk of infection.
The criteria is
Progressive increase of head circumference.
Increase soft spot tension.
Widening of skull sutures.
After the clinical evidence summarized above I request a brain CT scan. I pay attention to the size of the ventricles but also to the frontal subarachnoid space, and the basal cisterns. If the cisterns are wide similarly to the subarachnoid space I defer placing the shunt.
As we follow our patients well into adolescence we have observed that the psychomotor development of those who have not been shunted has not been impaired.
If the patient was not shunted I place an external ventricular drain. We sample CSF every 48 hours. After 14 days and according to cytology and growth we will either,
Replace the draining system, assuming it is colonized,
Or place the definitive ventricular shunt
Or consider that the patient does not need the shunt.
Chiari II
Of the 4 types of Chiari malformation, type II is associated with myelomeningocele.
There is radiological evidence in 80% to 90% of patients. Of those, only 20% manifest with symptoms.
Symptoms are related to brainstem function, either secondary to compression or to malformation.
When symptoms presented at birth that suggest a bad prognosis.
Inspiratory stridor
Swallowing difficulties that leads to aspiration pneumonia
Cyanosis an apnea.
Those symptoms are directly related to brain stem malfunction.
En Guatemala, la prevalencia de anomalías del tubo neural, es de aproximadamente 2.34 por 1,000 nacidos vivos. Y representan las anomalías externas más frecuentes, más que las de la pared abdominal y las de labio leporino.
Si tomamos en cuenta que el país tiene un crecimiento anual cercano al 2.8%, significaría que nacen anualmente 336,000 niños. Esto sería aproximadamente 786 niños y niñas con anomalías de este tipo al año en toda la República.
La distribución geográfica de las Anomalías del Tubo Neural, indica que la prevalencia es mayor en el noroccidente del país, donde existe una alta concentración de población indígena y los peores indicadores de situación nutricional.
De alguna manera, algunas investigaciones orientan a que en la etiología de estas anomalías en Guatemala, pueden asociarse a la deficiencia de ácido fólico, al consumo de micotoxinas que impiden la captación celular de àcido fólico (fumonisinas) o bien a algunos trastornos donde participa un gen, involucrado en el metabolismo de la tetrahidrofolato reductasa, enzima clave en el aprovechamiento del àcido fólico fisiológicamente activo.
Entre ellas tenemos: Anencefalia
Espina Bífida Abierta y Cerrada.
Encefaloceles Anteriores y Posteriores.
Con esto concluimos que, la espina bífida es la malformación congénita del sistema nervioso central más común y entre ellas la forma abierta representada por el Mielomeningocele es la más frecuente.
Si bien se sabe por estudios internacionales, que son prevenibles en un 50 a 70% con la ingesta de ácido fólico, se considera que por la gran incidencia que presenta en este país, hay también una causa genética, ya que se presentan varios casos en una misma familia, así como también se puede recabar antecedentes familiares en grados más lejanos en un 30% de los casos, los cuales no viven en la misma localidad.
Por tal razón, desde que llegué desde mi tierra natal, Argentina, visualicé la alta frecuencia de la Espina Bífida, la cual debía ser atendida en forma integral y multidisciplinaria por lo que logré crear una Unidad de Espina Bífida e Hidrocefalia y otras Anomalías del Tubo Neural, en el Hospital General San Juan de Dios, la cual en la actualidad funciona como única Unidad de referencia de toda la República e incluso de Centroamérica.
En esta Unidad realizamos más de 250 cirugías anuales, entre las 65 a 70 son cierre de Mielomeningocele.
En Guatemala existen barreras para la atención adecuada de los pacientes con EB, entre las que figuran culturales, socioeconómicas, geográficas.
Aunado a esas barreras se encontraba el estereotipo de que estos pacientes serían retrasados mentales y paralíticos, de ese modo se los trataba tardíamente o no se hacía nada por ellos, con una notoria indiferencia en vez de verlos como un problema de salud a nivel nacional.
El Mielomeningocele es la forma de Espina Bífida más frecuente y más grave, es una anomalía compleja, no solo afecta al Sistema Nerviosos Central, sino al genitourinario y al locomotor, por lo tanto se asocia con agenesia o hipoplasia renal, hidronefrosis, hidroureter, reflujo vesico uretral, a problemas de columna (cifosis o cifoescoliosis) deformidades del pie (equinovaro) y luxación de caderas.
Se describe la mayoría de las veces, como una bolsa en la espalda de contenido líquido, aunque otras veces es plana.
Tiene una parte central que representa a la médula expuesta, llamada PLACA NEURAL de color rojo, rodeada de una membrana transparente de color azulado, que es la ARACNOIDES, seguido de la PIEL DE TRANSICIÓN, la cual es hiperpigmentada, delgada ya que tiene poco tejido celular subcutáneo, es poco vascularizada haciéndola inadecuada para el buen cierre.
Mucho son los condicionantes que pueden modificar el pronóstico de los pacientes con Mielomeningocele, así en este contexto se pueden considerar:
El diagnóstico prenatal
En nuestro medio el diagnóstico prenatal se hace por ultrasonido, aunque solo un 40% tiene acceso al mismo
Grupo de manejo de la embarazada con diagnóstico prenatal.
Con la intención de operar a los pacientes con Mielomeningocele dentro de las 12 horas de nacido y ofrecerle de ese modo la mejor oportunidad de vida, formé un grupo interesado en el manejo de la embarazada con diagnóstico prenatal, en coordinación con Obstetricia y su Sección de Ultrasonido e integrado además por Psicología, Genética y Neonatología.
En cuanto Obstetricia capta a la embarazada con diagnóstico de la anomalía, interviene la Psicóloga quien apuntala a la mamá y a la familia (cuando la hay) ya que muchas de estas pacientes son jóvenes madres solteras a quien la propia familia la deja sola, como llevando una culpabilidad.
Se trabaja sobre la aceptación del niño/a, se le informa que estará en contacto con un grupo que espera el nacimiento de su hijo para darle el tratamiento.
El Genetista identifica si la lesión es aislada o sindrómica, evaluando el pronóstico junto con Neonatología y Neurocirugía, se elabora un plan de prioridades de tratamiento, como en el caso del Síndrome de Jarcho-Levin que presenta fusión de la parilla costal y el paciente será oxígeno-dependiente, en el cual lo respiratorio es el problema principal que regirá el pronóstico [Figura 1].
Figure 1
Síndrome de Jarcho-Levin
Las ventajas de contar con el diagnóstico prenatal es el de comenzamos tempranamente con el apoyo psicológico a la madre, para la aceptación del niño/a; así como también clasificar a la anomalía, si es aislada o sindrómica y estar preparados para enfrentar los problemas de otros sistemas que puedan comprometer la vida.
Planificar el día y tipo de nacimiento
Con respecto al día de nacimiento, en nuestro medio es importante porque tenemos días asignados para uso de sala de operaciones y nos cuesta lograr operarlos durante el horario de emergencias, ya que todavía hay resistencia a considerarlo como tal.
El tipo de nacimiento que indicamos es la cesárea, ya que la bolsa del Mielomeningocele puede romperse en el parto y esa fístula lo convierte en EMERGENCIA ABSOLUTA, lo cual aumenta el riesgo de sepsis.
Si esta ruptura sucede en un Hospital Departamental, en donde no cuentan con Neurocirujanos, se les indica que le realicen una sutura simple para cerrar esa fístula antes del traslado a la capital.
La cirugía oportuna
La cirugía del cierre del Mielomeningocele, la realizo al nacer en cirugía continua inmediata a la cesárea o en las 12 horas de nacido.
Porque la cirugía temprana?
Lo más importante es evitar la infección del Sistema Nervioso Central, que es tan grave en el recién nacido y si sobrevive tendrá secuelas definitivas.
En el caso de los gérmenes gram negativos, producen zonas de encefalomalacia con formación de cavidades en todo el tejido cerebral.
La otra razón es evitar el deterioro neurológico progresivo
Hay que tener en cuenta que en los hospitales Nacionales las infecciones se presentan en un alto porcentaje y que el paciente con Mielomeningocele ingresa a una sala de Neonatología general, ya que no es considerado un recién nacido de alto riesgo.
En este medio no siempre lo puedo lograr, ya que la mayoría de los pacientes llegan por referencia de los Hospitales Nacionales que se encuentran en los Departamentos en donde solo le brindan atención primaria y los trasladan hacia la Capital, viajando durante 4 a 9 horas en ambulancia y al llegar suelen estar descompensados por deshidratación, hipotermia, acidosis.
El seguimiento por grupo multidisciplinario de especialistas
Es tan importante este seguimiento multidisciplinario que lo considero el otro 50% después de la cirugía en el tratamiento de estos pacientes.
Después de la operación, el paciente es citado al Consultorio Multidisciplinario de Espina Bífida e Hidrocefalia, en donde en un mismo día, es evaluado por 10 especialidades: Neurocirugía, Urología, Ortopedia, Cirugía de Columna, Pediatría, Fisiatría, Psicología, Cirugía Pediátrica, Estimulación Temprana y Trabajo Social. Para diagnóstico, control y tratamiento de:
Anomalías asociadas: Hidrocefalia
Chiari II sintomático
Anomalías secundarias: Vejiga neurogénica
Reflujo vesicoureteral,
Infecciones urinarias,
Incontinencia esfinteriana,
Deformidades de los pies,
Luxación de cadera
Cifoescoliosis,
Síndrome de médula anclada.
Las citas son personalizadas de acuerdo a cada paciente
El apoyo psicológico a los padres y la estimulación temprana al ñino/a.
Cuando nace un niño con Mielomeningocele se produce un impacto emocional, los padres pueden sentir en un primer momento un rechazo a ese niño/a que viene a cambiar su vida cotidiana, la familia siente esa desintegración, ya que la madre ahora tiene que quedarse con ese hijo en el Hospital por largos períodos, la cual a veces lo abandona.
La rehabilitación física permanente
La fisioterapia debe comenzar lo antes posible, al pasar el período del estrés post-quirúrgico y continuar con sesiones 2 a 3 veces por semana en un centro calificado
Tratamiento del Mielomeningocele
El protocolo de ingreso para la cirugía de urgencia es el siguiente:
Laboratorios prequirúrgicos de rutina
Posición del paciente en decúbito ventral
Radiografía de columna del nivel de la lesión para evaluar si hay cifosis y en qué grado se presenta para la conducta quirúrgica a seguir, ya que en los casos de cifosis severa angulada, realizo cifectomía o corpectomía en el momento del cierre del Mielomeningocele.
Antibióticos para Sistema Nerviosos Central, desde el nacimiento.
El protocolo de estudio ya en la sala de internación es:
Neuroquirúrgicos: Ultrasonido Transfontanelar,
Tomografía de cerebro solo si vamos a colocar una válvula.
Urológicos: Cultivo de orina, muestra por sondeo vesical.
Ultrasonido renal
Uretrocistograma
Ortopédicos: Rx de cadera en posición de rana
Rx de ambos pies en caso de deformidades
Rx de columna
Cirugía del Mielomeningocele. Como lo hago yo
Desde el punto de vista de la dificultad en el cierre de la piel y la conducta a seguir, clasifico al Mielomeningocele en Simple y en Complejo.
En el primer grupo sigo la técnica clásica y en el segundo, a su vez los divido en 3 grados. (I-II y III)
Cirugía AL NACER o dentro de las 12 horas de nacido
La razón principal es evitar la infección del SNC (Pioventriculitis) sumamente grave en el recién nacido, el cual tendrá que pasar por una larga estancia hospitalaria, sumándose el riesgo de adquirir infecciones nosocomiales y complicaciones como Neumonía y otras.
Otra es evitar el deterioro neurológico progresivo. Pasos de la Cirugía:
Despegar el sistema nervioso de la piel.
Hago la incisión con bisturí 15 en la línea que divide la piel de transición y la aracnoides, en posición lateral, completo con tijera Metzembaum o tijera pequeña que se usa para el iris, teniendo mucho cuidado en la línea media en donde habitualmente se encuentra el tejido medular pegado a la piel.
En este momento la placa neural rodeada de la aracnoides, separada de la piel de transición, “cae” al canal medular.
Para el control de la hemostasia utilizo pinzas pequeñas de hemostasia, llamadas “mosquito”, colocándolas en los bordes internos de la incisión, con mucho cuidado de no lesionar la piel, las cuales también me sirven para la separación, ya que los separadores automáticos con los que contamos son traumáticos para este tipo de tejido.
Tratamiento de la placa neural.
El segundo paso es extirpar la ARACNOIDES pegada al borde de la placa neural, lo hago utilizando en forma intermitente la coagulación bipolar y una tijera delicada, se va cortando lo coagulado para evitar la hemorragia, hasta que quede la placa neural con el borde libre.
Posteriormente hay que tomar la decisión de qué hacer con la placa Neural según su forma y grosor.
Si la PLACA NEURAL es pequeña y se introduce fácilmente en el saco dural no es necesario invaginarla.
En cambio cuando la Placa Neural es grande y gruesa, es necesario disminuir ese grosor, para lo que utilizo bisturí 15, y en forma horizontal “rebano” cuidadosamente
sobre la cara a superior, la que estuvo en contacto con el exterior, controlando la hemostasia con bipolar y lograr de ese modo invaginarla para darle la forma tubular y para no dejar una zona cruenta que pudiera adherirse posteriormente con más facilidad y anclar la médula.
Compruebo que no hay adherencias para la libre movilización de la médula.
Reconstrucción del saco dural:
La disección del plano dural, el cual se encuentra formando el piso de la lesión, lo hago con una pequeña incisión y lo completo con tijera.
El cierre lo hago con Prolene 4-0, puntos continuos, al final del mismo solicito a anestesia que hagan una maniobra de Valsalva para comprobar que no hay fuga de líquido cefalorraquídeo.
Tengo en cuenta que el cierre del saco dural no sea comprimiendo la médula y que no tenga adherencias, para evitar el anclaje medular.
Cierre de la piel sin tensión, esto lo considero fundamental, ya que
de no hacerlo, la consecuencia será una dehiscencia de la herida operatoria con el consecuente riesgo de infección.
Diseco el plano avascular por encima del músculo hasta 4 cm o más con coagulación monopolar, con lo que logro que la piel ceda hasta llegar a la línea media.
Coloco unos puntos de Vicryl 3-0 con aguja cortante, en el borde de tejido que se encuentra, en lo que yo le llamo “base de implantación del Mielomeningocele” en el ángulo que forman la piel de transición y la piel sana, con eso sostengo la tensión del cierre de la herida operatoria, evitando el cierre de la piel en forma tensa.
Finalmente recorto la piel de transición sobrante, la cual no utilizo para el cierre porque tiene poca vascularización y suturo los bordes de la piel sana sin tensión, con puntos continuos pasados o separados, según lo necesite con nylon 4-0.
La localización más frecuente es a nivel lumbosacro, pero existe un significativo porcentaje (30%) de localización dorsolumbar con Hidrocefalia severa y cifosis angulada, grandes lesiones en las que se dificulta el cierre de la piel sin tensión [Figuras 2 – 4 ].
Figure 2
Grado 2
Figure 3
Grado 3
Figure 4a
Pacientes en los que no se corrigió la cifosis angulada en el cierre del Mielomeningocele, con dehiscencia de la herida operatoria
Figure 4b
Dehiscencia de herida operatoria en Mielomeningocele con cifosis
Mielomeningocele Complejo
Se subdivide en 3 grados (Mannucci)
El momento de la Cirugia lo hago diferido a la primera semana de vida.
Grado 1
Es el MMC que presenta base ancha de impantación, por lo tanto dificultad en el cierre de la piel.
Para lo que utilizo incisiones de descarga laterales, técnica de Cirugía Plástica más sencilla que las descritas en los libros con grandes incisiones en Z u otras de rotación de los músculos del dorsales.
Grado 2
Es el que presenta cifosis angulada, en estos casos realizo la técnica de la cifectomía o corpectomía en el momento de cierre del MMC, para lograr el cierre de la piel sin tensión y para corregir la cifosis, la cual será progresiva, con sus respectivas consecuencias posteriores en la calidad de vida.
La técnica se realiza en forma convencional hasta el cierre del saco dural, posteriormente se identifica el espacio intervertebral y se extrae el cuerpo identificado en el ángulo de la cifosis, personalmente la mayoría de las veces es de solo un cuerpo, posteriormente con puntos de seda 2-0 se afrontan las partes blandas de los bordes en donde se encontraba el cuerpo vertebral y de ese modo, disminuye la angulación y se cierra la piel sin tensión.
Grado 3
Es en el que se suman los dos anteriores base ancha de implantación y cifosis angulada, el más severo en gravedad, cirugía más prolongada, via central, transfusión sanguínea, cirugía prolongada, por lo que se realiza después que el recién nacido se ha estabilizado, pero antes de que se infecte, por lo que recibe cuidados intensivos.
Cifosis angulada progresiva en paciente que no se corrigió la cifosis en la cirugía de cierre del Mielomeningocele.
La cifosis progresa, si no se corrige desde la Qx del cierre crece en forma angulada y traerá consecuencias posteriores invalidantes como es la disminución de la capacidad pulmonar.
La Hidrocefalia está presente en el 80% de los casos, sin embargo solo se evidencia al nacer en un 20%.
La colocación de la válvula la hago solamente si compruebo la que la Hidrocefalia es progresiva, es entónces cuando pido una TAC de cerebro y según la tabla de Riesgo de Sepsis que manejamos en la Unidad (grados I, II Y III), indico punción ventricular para comprobar que no hay infección mediante el exámen químico, citológico y el cultivo del LCR.
En mi experiencia, he podido comprobar, coincidiendo con el Profesor Maurice Choux, Jefe de Neurocirugía del Hospital de Niños La Timone, Marsella, a quien tuve el gusto de escuchar Congresos Latinoamericanos de Neurocirugía, que esta forma de Hidrocefalia que acompaña al Mielomeningocele se detiene en el 30% de los casos, no necesita válvula de derivación y el paciente presenta un desarrollo psicomotor normal.
En ningún caso coloco la válvula de derivación en el mismo acto operatorio del cierre de MMC, ya que prolonga innecesariamente el acto operatorio y aumenta el riesgo de infección.
Ventriculomegalia visualizada en la Tomografía de cerebro, no es lo único que indica la operación
Se realiza ultrasonido transfontanelar al nacer y se controla en forma estricta los signos clínicos de Hidrocefalia progresiva:
Aumento anormal del perímetro cefálico, medición diaria.
Tensión y amplitud de la fontanela anterior
Crecimiento de la diastasis de suturas, principalmente evidenciada en la sutura coronal.
La TAC de cerebro se indica en los casos en los cuales se presenten los signos anteriores
Tomografía computada de cerebro: en este estudio tengo en cuenta además de la ventriculomegalia otros signos como son la visualización de las cisternas (de la base, silvianas, interhemisférica) y el espacio subaracnoideo frontal.
En casos de macrocefalia y dilatación ventricular en la tomografía computada de cerebro, si veo que son amplias las cisternas y el espacio subaracnoideo frontal, y que los signos los signos clínicos son “lentamente” progresivos o se estacionan, no coloco la válvula en ese momento, sino entra en un control evolutivo estricto por consulta externa para evaluar el comportamiento de esa Hidrocefalia.
En la Unidad de espina Bífida e Hidrocefalia se da seguimiento a los pacientes hasta la adolescencia, pudiéndose comprobar el desarrollo psicomotor normal en estos pacientes en los cuales no se les colocó la válvula.
Hidrocefalia infectada
En caso de infección del LCR en una Hidrocefalia progresiva, coloco un sistema cerrado de ventriculostomía, el cual se mantendrá durante 14 días, tomando muestras de LCR cada 48 horas y evaluar cambios químicos y citológicos, que nos indiquen si hay mejoría con el tratamiento antibiótico instaurado por antibiograma, en caso de que no mejore, plantearse el cambio del mismo.
El cultivo puede dar un falso negativo por la antibioticoterapia.
Cuando llega el día 14, nos hacemos dos preguntas, para la conducta a seguir:
Está resuelta la infección?
Es el paciente válvulo-dependiente?
Si está resuelta la infección y el paciente es válvulo dependiente, se le coloca
Una válvula de derivación ventrículo peritoneal.
Si está resuelta la infección y no es válvulo dependiente, se retira la ventriculostomía y se da egreso.
Si no está resuelta la infección se cambia el sistema cerrado de ventriculostomía, ya que si a 14 días no ha resuelto, se considera que el catéter está colonizado.
Chiari II
La malformación de Chiari, que se define como un descenso de las amígdalas cerebelosas en el canal medular tiene 4 tipos, de los cuales el tipo II se asocia a Mielomeningocele e Hidrocefalia por lo que estas dos patologías dominan el cuadro clínico el cual se presenta como disfunción del tronco y de los últimos cuatro pares craneanos.
La mayoría de los pacientes la presentan, se habla de 80 a 90% de los mismos, pero hay formas asintomáticas y sintomáticas.
Hay que tener en cuenta que el tronco encefálico es un tronco malformado, que a su vez se encuentra descendido y a su vez comprimido por el cerebelo.
Si la sintomatología comienza de recién nacido es un mal signo, como son:
Los episodios de estridor inspiratorio (paresia del x par) y apnea con cianosis.
Trastornos deglutorios, lo que provoca neumonías aspirativas y regurgitación.
Llanto débil
Estos síntomas debe tenerse en cuenta que son causados en forma directa por la malformación y de mucha gravedad.
Se considera que solo un 20% de los pacientes tienen Chiari sintomático
La forma de presentación del niño en la segunda infancia es diferente a la del recién nacido
Ya que se caracteriza también por síntomas de los 4 últimos pares craneanos, pero también hay., signos Cerebelosos (ataxia, asinergia, nistagmos)
dolor cervical que con los movimientos en flexión del cuello aumenta
compresión de vías largas del tronco, signos de hipertensión endocraneana por hidrocefalia;sintomatología más lenta e insidiosa no con la gravedad del neonato.
Para indicación de Cirugía descompresiva me baso en tres parámetros:
La cínica antes descrita
La imagen de la Resonancia Magnética de la unión craneocervical.
Estudios neurofisiológicos; Polisomnografía, Potenciales evocados de tronco.
Antes de realizar la descompresiva de fosa posterior, se debe de tener seguridad del funcionamiento de la válvula de derivación ventrículo peritoneal, que se colocó anteriormente por Hidrocefalia.
Medula anclada post-cirugía del Mielomeningocele
Hay que diferenciar la médula anclada, del síndrome de médula anclada, ya que todos los pacientes operados de mielomeningocele tienen la médula descendida y cierto grado de adherencias alrededor de las estructuras nerviosas.
Por lo tanto el diagnóstico es clínico y son los siguientes:
Pie cavo progresivo
Escoliosis progresiva
Paraparesia
En niños más grandes dolor en la parte posterior de las piernas.
Desmejoramiento en la continencia esfinteriana.
El objetivo de la cirugía es liberar la médula espinal y sus raíces de la cicatriz quirúrgica y cerrar la duramadre con la suficiente amplitud para que no vuelva a anclarse.
Técnica quirúrgica
Incisión mediana
Laminectomía de un nivel superior para la identificación de la duramadre sana.
Abertura la duramadre caudalmente y lateralmente a la cicatriz
Departamento de Neurocirugia, Hospital de Niños Ricardo Gutierrez, Ciudad Autonoma de Buenos Aires, Argentina
Correspondence Address: Santiago Portillo Departamento de Neurocirugia, Hospital de Niños Ricardo Gutierrez, Ciudad Autonoma de Buenos Aires, Argentina
Cord lipomas (CL) are another form of spina bifida. They are covered by skin and are less frequent than myelomeningoceles (MMCL) [Figure 1].
Figure 1
Cord lipoma covered by skin
It is a fatty tumor that frequently is in continuity with the subcutaneous adipose layer. Some times through a raquischisis attaches to the cord and even infiltrates it [Figure 2].
Figure 2
Cord lipoma, with perianal anomalies
Anatomically a CL resembles an intramedullary tumor albeit its symptoms are secondary to the tethering of the cord as it is an axis that connects the cord with the subcutaneous tissue.
Many patients come to the consult without any symptoms. My experience has thought me that eventually the majority will develop some form of neurological or urological impairment.
There is controversy regarding the indications for preventive treatment. The French School favors operating only when there are symptoms related to tethering of the cord.
The association between CL and perianal malformations has been described having been observed between 1.8% to 5.1% of patients with CL. In a retrospective study at the Department of Neurosurgery Hospital de Ninos Ricardo Gutierrez we observed that over 82 patients with CL 4 (5%) presented perianal malformation. The clinical picture was a combination of motor deficit, with neurogenic bladder, anal malformation, urinary bladder exstrophy and vertebral anomalies (hemi vertebra and total or partial sacral agenesis).
The embryological deficit is related to the process known as secondary neurulation. This process involves a mass of pluripotential cells that gives origin to the caudal part of the spinal cord and involves the development of vertebra and viscera as well [Figure 2].
Somato sensory evoked potentials and Electromyography of the lower limbs allows for preoperative assessment of the level of the lesion and for transoperative monitoring.
It is to reduce the mass of the tissue and to detach it from the extradural planes.
Skin incision
In filum lipomas we choose a midline incision. In transitional we opt for surrounding the dome of the lipoma, in this fashion we are immediately at the muscle plane and from there we identify the stem of the lesion.
Laminectomy
In filum lipomas we remove two laminae. In transitional lipomas we take advantage of the defect and from there we go two levels above the lesion so we can start working from normal cord towards the lipoma.
Dural plane and lipoma
We work under microscope. The objective is to release the mass avoiding damage to the cord and the roots.
In transitional and dorsal lipomas I utilize bipolar coagulation and I am constantly aware of the relation of the mass with the underlying cord. I keep in mind that the objective is to reduce the mass.
In the case of terminal lipomas, once we identified the fatty filum, determine that there are no rootlets adhered to it and proceed to divide it.
At the time of closing dura I keep in mind that the objective at this stage is to avoid tethering of the cord thus if required I enlarge the dura sac with a patch of fascia.
I personally favor preventive surgery. Our series at the Hospital de Niρos evidenced that on immediate postoperative period 2.5% had worsened motor function and 8.5% had worsened bladder and sphincter function. Longtime assessment evidenced that only 6% of patients had sequelae from the surgery, mostly related to bladder and sphincter function.
I am respectful of those authors who prefer to operate only after clinical evidence of tethered cord syndrome. They base their argument on that surgery increases the risk of long term tethering of the cord secondary to postoperative adhesions.
In our experience, the long term follow up of patients younger than one year have not evidenced symptoms that would have required a new procedure. The age of the patients is significant because those very young are the ones more prone to evidence rethetering because of longitudinal growth of the spinal canal.
We have improved the surgical technique to reduce the chances of arachnoid adhesions.
In a great number of transitional lipomas there is no clear boundary between fat and nervous tissue. Our aim is to reduce the mass and not total removal. We limit our work to the level of entry of the dorsal roots. The ganglions of the dorsal roots originate from the external layer of the neural tube immediately adjacent to the level at which the neural tube folds into its final shape.
Los lipomas medulares constituyen otra forma de espina bífida cubierta por piel, que no son tan frecuentes como el mielomeningocele (Figura 1).
Figura 1
Lipomas medulares como lesión cubierta por piel
Consiste en una tumoración grasa que la mayor parte de las veces se encuentra en contacto con el tejido graso subcutáneo. A través de una raquisquisis y un defecto en las cubiertas meníngeas se conecta con la médula con quien toma una relación hasta cierto punto “infiltrativa”. Su comportamiento anatómico recuerda el de los tumores intramedulares, sin embargo su sintomatología corresponde al anclaje medular, dado que constituyen masas de tejido conectivo que están definitivamente en contacto con la médula y fijan a esta con la meninge y con el tejido celular subcutáneo circundante.
Muchos pacientes que consultan por lipomas medulares son neurológicamente asintomáticos al momento de la consulta, sin embargo mi experiencia, muestra que con el paso del tiempo, la mayoría, presentarán algún tipo de deterioro neurológico o urológico.
El tratamiento quirúrgico de estos pacientes, sobre todo los que se encuentran asintomáticos es controversial, debido a las secuelas, y complicaciones que puede ocasionar el procedimiento quirúrgico. Por ello algunos como la Escuela Neuroquirúrgica Francesa adoptan una conducta expectante con seguimiento clínico, recurriendo a la cirugía solo si hay sintomatología atribuible al anclaje medular.
La asociación de Lipomas medulares con malformaciones perineales ha sido descripta con una frecuencia que varía entre un 1.8% al 5.1%. En un estudio retrospectivo realizado en el Servicio de Neurocirugía del Hospital de Niños Ricardo Gutiérrez, sobre 82 pacientes con diagnóstico de lipoma medular, 4 pacientes (5%) presentaban este tipo de asociación. Este cuadro se conforma por la existencia de tres o más características: síndrome cutáneo, síndrome neurológico (alteraciones motoras de miembros inferiores y/o vejiga neurogénica), síndrome perineal (malformaciones ano rectales, extrofia vesical) y anomalías vertebrales (fundamentalmente hemivértebras y agenesia parcial o total sacra).
La explicación embriológica probable de esta conjunción de malformaciones y su relación con la médula espinal está dada en la denominada “segunda neurulación” que tiene que ver con la “eminencia caudal”, que es una masa de tejidos pluripotenciales que dan origen a la parte más caudal de la médula espinal, células de la cresta neural, notocorda caudal (vértebras sacras), mesénquima caudal, somitas caudales, et. De está forma cualquier trastorno en este proceso, se puede traducir en defectos que en menor o mayor medida involucraran al cono medular, a las vértebras sacras, al intestino y a la región perineal [Figura 2].
Figura 2
Lipoma medular con la combinacion de miembro accesorio “Parecido a un pie” y alteraciones perineales
Esta clasificación se basa según las relaciones del tejido graso con el tejido nervioso de la médula
De esta forma se clasifican en:
Lipomas de Filum
Lipomas Transicionales
Lipomas Dorsales
Lipoma de Filum
Son aquellos que se insertan en la parte terminal del cono medular sin mezclarse con él ni con la región de ingreso de las raíces dorsales
Lipoma Dorsal
Se insertan y algunas veces mezclan con la porción dorsal de la médula espinal pero a nivel segmentario.
Lipoma Transicional
El lipoma se inserta y mezcla con la porción más distal de la médula (el cono medular), no existiendo por ello más médula normal por debajo de la inserción del lipoma
Tanto los lipomas dorsales como los transicionales comparten la característica de que el tejido graso que deja la médula se une a su vez con el tejido graso subcutáneo.
En orden de tratar de zanjar la controversia sobre la evolutividad de los síntomas y la necesidad de tratamiento quirúrgico hicimos un estudio retrospectivo en un periodo de 22 años entre 1988 y 2010, encontrando 32 pacientes mayores de 3 años y 50 menores de 3 años al momento de la primera consulta. En los menores de 3 años el motivo de consulta más frecuente (85%) era la tumoración lumbo sacra, mientras que en mayores de tres años el 42% tenía síntomas de disfunción urológica o trastornos motores y solo el 24% acudía a la consulta por la tumoración. Ello demuestra la evolutividad de la sintomatología.
En mi experiencia los síntomas más frecuentes son:
Trastornos motores de miembros inferiores: alteraciones en la posición de apoyo de los pies, atrofias musculares, contracturas y trastornos tróficos por falta de sensibilidad en el área afectada. Esto lleva a la sintomatología referida por el paciente: dificultad progresiva en la marcha
Escoliosis: tanto relacionada a las malformaciones vertebrales presentes, como al anclaje medular que se produce por el Lipoma.
Tomografía computada de columna: Permite conocer la presencia de grasa a nivel del canal medular, y las probables malformaciones óseas vertebrales; pero no permite definir la relación médula – grasa, ni la extensión de la grasa dentro del canal.
Resonancia Magnética por Imágenes de Columna: Es el estudio ideal porque permite tener una idea en los tres planos espaciales, de la relación de la grasa con la médula, lo que permite establecer la estrategia quirúrgica adecuada e incluso conocer de antemano el tipo de Lipoma
Estudios Neurofisiológicos
Somatosensitivos y Electromiograma de miembros inferiores: son de gran utilidad en la definición del nivel lesional presente y en el monitoreo intra quirúrgico en aquellos casos que re-anclaje medular.
Es liberar la médula de la adherencia que representa la grasa dentro del canal medular a la ubicada en el plano subcutáneo, además de reducir al mínimo la grasa que se encuentra infiltrando el cono medular
Incisión
Utilizamos una incisión sobre la línea media posterior lumbar en el caso de los lipomas de filum terminal. En el caso de los Lipomas transicionales de cono con grandes tumores grasos subcutáneos, usamos una incisión que bordea al tumor o sea es paravertebral curva. Este tipo de incisión nos permite alcanzar rápidamente el plano muscular y desde ahí rodear la grasa estableciendo claramente el defecto a través del cual de subcutánea pasa a intra-canalicular; por otro lado nos permite reducir el tumor sin desvitalizar la piel.
Laminotomias
En el caso de los lipomas de filum usamos una o dos Laminotomias sobre el lugar donde pensamos que ya no hay raíces para poder abrir la duramadre y seccionar el filum lipomatoso. En el caso de los lipomas transicionales del cono aprovechamos las malformaciones vertebrales y si no una o dos Laminotomias por encima del defecto de forma de comenzar a trabajar en el plano intradural desde el tejido sano
Plano dural y Lipoma
Se utiliza el microscopio quirúrgico desde la apertura dural, considerando que la misma debe extenderse hasta visualizar médula sana, por encima de la lesión lipomatosa, identificando las raíces nerviosas, para no lesionarlas, hasta cumplir el objetivo fundamental que es liberar la médula de sus adherencias durales, y de la fijación que le impone el lipoma subcutáneo.
En los lipomas transicionales y dorsales se procede a la exéresis del lipoma sin intentar una remoción total, utilizando la coagulación bipolar y teniendo en cuenta las relaciones anatómicas de la interfase lipoma-tejido nervioso y raíces nerviosas.
En el caso de los lipomas terminales (figura A, B, C, D), una vez que el filum lipomatoso se identifica, se separa de adherencias a las raíces nerviosas, se secciona y se extrae en el límite con el tejido nervioso, siempre y cuando éste se pueda identificar. Finalizado esto, se procede a la reconstrucción de un saco dural amplio. En el caso de necesitar un parche de duramadre la misma se hace con fascia muscular, de forma tal de dejar un saco amplio, que impida el re anclaje.
Soy proclive a la cirugía del lipoma aún en pacientes que son asintomáticos. Los resultados de la serie del Hospital de Niños Ricardo Gutiérrez muestran que este procedimiento puede ser realizado con un margen aceptable de seguridad, en el post operatorio inmediato sobre 82 pacientes solo un 2,5% habían empeorado la función motora, en cambio el 8,5% habían empeorado la función urológica, a largo plazo lo único que persistió fue un 6% con secuelas urológicas. Sin embargo consideramos aceptable, lo propuesto por los autores, que sugieren una conducta expectante en relación a los pacientes con lipomas transicionales sin síntomas. Ellos consideran que hay posibilidad de que el deterioro neurológico no sea inmediato, por daño en estructuras neurológicas, sino tardío como consecuencia de adherencias a la cicatriz (síndrome de médula anclada). Estos autores refieren que el deterioro neurológico se evidencia en un 50% a largo plazo y no en el post - operatorio inmediato, atribuyendo este fenómeno a adherencias quirúrgicas. Tenemos seguimiento a largo plazo de los pacientes de menos de un año de edad que son los que afrontarán más los mecanismos fisiológicos de producción de la médula anclada: mayor crecimiento de la columna vertebral, que de la médula espinal, y por ende mayor exposición a la tracción por adherencias a la cicatriz, en ellos no hemos visto deterioro motor que requiera cirugías para nuevo desanclaje medular.
Hemos depurado la técnica quirúrgica para realizar una reconstrucción de los planos aracnoidales en la búsqueda de disminuir la superficie cruenta que pudiera generar posteriores adherencias a la cicatriz.
En la mayoría de los lipomas transicionales el límite tejido nervioso - lipoma, no es muy claro, por lo cual consideramos que el éxito de esta cirugía debe basarse en privilegiar la liberación medular sobre la exéresis “total” del lipoma. Debe realizarse una descompresión medular. Sin embargo resulta claro que la entrada de las raíces dorsales a la médula espinal, marca el límite más externo del lipoma, puesto que los ganglios de las raíces dorsales se originan a partir de células de las crestas neurales, las cuáles a su vez se originan de la capa más externa del tubo neural, inmediatamente adyacente al punto donde se fusionan los pliegues neurales para constituir el tubo neural.
Finalmente, consideramos que es necesario un adecuado entendimiento de la embriología de este tipo de defectos congénitos antes de encarar la táctica quirúrgica, debido a la gran variabilidad anatómica de los mismos.
Está descripto el empeoramiento del cuadro clínico previo. En nuestra experiencia esto ocurre pocas veces y en forma transitoria.
Quizás la complicación más importante y más frecuente sea la fístula de líquido céfalo raquídeo a través de la herida quirúrgica. Ella puede obligar a otra cirugía para cerrar la brecha dural, o incluso a colocar un drenaje subaracnoideo al exterior de LCR por encima de la herida quirúrgica a fin de permitir la cicatrización de la misma.
While alpha-fetoprotein is indicative of an open neural tube defect, 40% of patients seen at our clinic in Argentina have had a diagnostic ultrasound. Majority of those patients had the study in the last trimester even on the week previous to delivery.
Not always the child has the typical sac filled with fluid that simplifies the diagnosis while looking at an ultrasound, thus the incidence of smaller lesions and even flat lesions may account for the cases in which the parents were not aware of the condition of their child.
In Argentina we have seen a decrease of the large thoracic lesions. This could be secondary to the implementation in 1993 of the law that mandates folic acid fortification of the flour.
That in spite of this measure we still have cases suggests that physicians should be alerted on the value of recommending oral supplement of folic acid before pregnancy and also that not all the cases are necessarily related to folic acid deficit.
Free the placode from the lateral adhesions. Be extremely careful when detaching the placode cephalically and caudally. To avoid damaging the normal cord and, caudally, to preserve the integrity of the cauda equina.
Remove vestiges of dermal tissue to prevent growth of dermal cysts.
Oppose the lips of the placode to restore the cylindrical shape of the cord
Dissect, flap over and suture dura over the placode.
Dissect and flap muscle fascia. There is controversy about this maneuver. I our experience there is no increased incidence of kyphosis or scoliosis and furthermore, our physical therapists have noticed that when manipulating the lumbar area in those children without a muscle layer over the dura plane it triggers involuntary movements.
Adequate closure of skin. Release skin incision if necessary. We prefer “z” shaped. Always considering that a new surgery may be required for stabilizing the column.
Explore the canal cephalically with the handle of a Penfield or similar small and round instrument for diastomatomyelia that could be repaired.
Kyphosis. During my training we never contemplated corpectomy because of its high morbidity. Now with advance anesthetic and perioperative care this is possible. And still, I am weary of performing corpectomy because we are transforming a stable column into an unstable one. This delays the rehabilitation and does not address the associated problem, frequently seen, of a malformed thorax that affects the quality of life of the child. I don’t think that early corpectomy is advantageous for the child.
The objective of the surgery is to
Prevent infection by covering the exposed tissue and repairing the cerebro spinal fluid (CSF) fistula.
Preserve neurological function. It goes without saying that while we completely agree on that the placode is already damage by its intra uterus exposure, it is very reasonable to presume that function will be further impaired if the wound is not carefully closed.
Imperfect closure of skin that may force the surgeon to help healing by granulation rather than re operate on a tissue that has already proven to be poorly vascularized to begin with.
Present in 95% of the cases. The shape of the lateral ventricles is particular to the children with MMCL due to associated malformations such as massa intermedia, dysgenesis of the corpus callosum.
Endoscopy has allowed us to visualize the lateral ventricles and has allowed us to observe that the large massa intermedia may block the Monro foramen leading to the collapse of one ventricle after shunting with subsequent entrapment of the III ventricle or the contralateral ventricle. We have also seen defects in the ventricular septum, larger choroid plexus than in patients without MMCL and ventricular wall heterotopy.
Hydrocephalus is secondary to
Stenosis of the aqueduct
Blockage at the level of the foramen of Luschka and Magendie.
Impaired subarachnoid flow related to thickened arachnoid.
Signs and symptoms of hydrocephalus
Progressive enlargement of the head circumference.
Worsening of symptoms associated with Chiari II (stridor, apnea, ophistotonos)
There are asymptomatic patients with radiological evidence of venticulomegaly. And here is when the surgeons experience is valuable.
Imaging
Ultrasound. Non-invasive, can be performed at the bedside. Informative
Head CT scan. Radiation. Accurate.
MRI. Ideal, caveat: requires anesthesia.
Treatment
Ventriculo Peritoneal Shunt. Usually in the first two months of life. If we wait longer we risk having a large skull thus increasing the risk of post shunt subdural collection.
We not necessarily shunt the contralateral ventricle if trapped.
Endoscopy. Not indicated, in my opinion, because the anatomy is already abnormal thus difficulting a safe procedure and more importantly the subarachnoid space is also anatomically altered thus flow along it may be impaired.
Shunt failure. 32% of our patients. Usually secondary to obstruction of the ventricular catheter by reactive glial tissue. Less frequent, mechanical issues such as disconnection or shorter catheter.
Shunt infection 17%. Before diagnosing infection of the shunt keep in mind that UTI is the most common cause of fever in children with MMCL. Keep in mind infection even when considering a shunt failure, particularly if the child has a loculated abdominal cyst that while giving symptoms of intracranial pressure it may be secondary to a bacterial infection.
The treatment is removal of the entire shunt system and placement of external ventricular drain or repeated ventricular tap through the patent anterior soft spot.
Staphylococcus Epidermidis is the most common pathogen.
In cases of ventriculo atrial shunt, the infection can cause shunt nephritis (staphylococcus coagulase negative) or in more severe cases by bacterial endocarditis with vegetation growth in the right chamber valves.
Chairi type II
Current definition is more complex than the one originally proposed by Chiari in 1896. Today we understand that the tonsils, the vermis, the brain stem and the IV ventricle are misplaced in the posterior fossa.
The vermis is abnormally anchored to the brain stem; this distorts the flow of CSF.
The tentorium is steeper.
Whereas almost every child with MMCL will have an anatomical Chiari II malformation, not everybody will present symptoms.
The symptoms are related to cerebellar and brain stem functions
Stridor, secondary to vocal cord palsy. The patient may be sub-cyanotic.
Apnea. It could lead to cardiac arrest of significant bradychardia.
Swallowing difficulties, expressed as repeated bronchial or pulmonary infection.
Weakness in the upper limbs.
Weakness of the trunk
Nistagmus. This usually associated to severe cerebellar malformation. Sometimes labeled as Chiari type IV.
Diagnostic imaging
Neuro axis CT scans.
Evidences absence of the cisterna magna.
Protrusion of the cerebellar tonsils through the tentorium.
Small or absent IV ventricle.
MRI. Is the preferred study. Allows for adequate assessment of the medulla
Neurophysiological testing
Auditory and somato sensory evoked potentials. What is considered is the delay in the transmission of the stimulus to the cerebral cortex and the shape of the wave.
Sleep study. To detect sleep apnea. The etiology and duration of the episodes and if they are accompanied by bradychardia.
Treatment
If apnea, stridor, swallowing difficulties.
Abnormal neurophysiological studies
Indicated before correcting tethered cord, scoliosis, and syringomyelia.
In the past we performed a duroplasty. Today we limit to cervical laminectomy.
The duramater has numerous venous lakes that may lead to massive hemorrhage. Manipulation of the brain stem may lead to significant bradychardia and increased arachnoid scarring.
Respiratory impairment secondary to brain stem malformation and surgical trauma.
Intense neck pain secondary to surgical incision.
CSF fistula if duramater was incised, accidentally or as a part of the procedure.
Distant complication
Reosification and reappearance of symptoms related to medullary compression.
Syringomyelia
We do not differentiate between syringomyelia and hydromyelia. Both are cystic dilatation of the medulla or cervical cord.
Syringomyelia is not evident at birth, perhaps because the CSF flows out of the central canal through the congenital defect.
As the child already has neurological deficit is not always possible to establish that the symptoms are related to syringomyelia. We value scoliosis as a red flag, as well as exacerbation of lower brain stem malfunction traditionally attributed to Chiari II.
MRI is the ideal study, superior to CT scan. (Figure).
Treatment
At the Ricardo Gutierrez Children's Hospital we opt for shunting the syrinx to the peritoneum.
We believe that syringo-arachnoid shunt fails due to congenital anomalies of the arachnoid and due to skeletal anomalies and finally CNS anomalies that conspire against the adequate flow and absorption of the fluid.
We did not have good experience with pleural shunts. The abnormal rib cage and the often already compromised respiratory function had cautioned us against syringe pleural shunts.
At our hospital we coincide with Harold Hoffman on performing a posterior fossa decompression first and then shunting the syrinx to prevent the complications that could arise due to the further descend of the cerebellum after releasing of intra-spinal pressure.
El mielomeningocele constituye el defecto del cierre del tubo neural más frecuente y compatible con la vida. A su vez es el que más morbilidad genera.
Si bien ya Aristóteles e Hipócrates habían hecho descripciones de esta patología, las posibilidades de tratamiento médico y por ende de sobrevida eran casi nulas.
Ha sido solo hacia fines de 1800 cuando se realizaron mejoras en la cirugía del cierre de este defecto y en 1950 cuando se colocaron las primeras prótesis valvulares para el tratamiento de la hidrocefalia, cuando se pudo obtener una mejoría en la morbimortalidad.
Si bien existen varios tipos de mielomeningocele, la descripción del más común, bastará para entender la importancia de un adecuado tratamiento quirúrgico inicial.
La deformidad más frecuente, consiste en una «placa neural abierta», la cual en general representa el extremo distal de la médula espinal. Este tejido nervioso se encuentra en contacto con piel normal. Sin embargo, un análisis más profundo, demuestra que existe una zona intermedia, denominada epitelio de transición, caracterizado por una fina membrana, que no es regular y puede no existir en algunas zonas en las cuales el tejido nervioso está directamente en contacto con la piel. Subyacente a la placa neural se encuentra el saco aracnoidal, en el que se encuentran las raíces nerviosas que salen de la médula espinal, y el líquido céfalo raquídeo. Hay que mencionar que la parte de la placa neural en contacto con el exterior, es la que al unir sus extremos laterales constituirá el interior de la médula, en tanto la que se encuentra en contacto con el saco aracnoidal es la cara externa de la médula. Delimitando el saco neural se encuentra la duramadre (Meninges).
Si bien se ha mencionado que este defecto puede ser detectado por la elevación de alfa feto proteína durante el embarazo, nuestra realidad nos indica que la mayor parte de los diagnósticos pre natales se hacen por ecografías.
En nuestro Consultorio Multidisciplinario donde recibimos pacientes con mielomeningocele de toda la Argentina, el diagnóstico pre natal se ha hecho solamente en el 40% de los pacientes, en la mayoría de ellos durante el tercer trimestre de la gestación y en algunos casos la semana previa al parto.
Pienso que esta falta de diagnóstico temprano obedece a que la mayor parte de defectos son defectos planos situados al mismo nivel que la piel, y no lesiones globulosas como las que los libros de Embriología o Diagnóstico por Imágenes pretenden enseñar como el “Mielomeningocele típico”, con lo cual la mayor parte de estudios de Ecografía pre natal se dirigen a verificar la vitalidad fetal y raramente buscan la columna o los signos precoces de hidrocefalia (signo del limón) o de Malformación de Chiari (signo de la banana)
En mi experiencia, me atrevería decir que desde la segunda parte de la década de los 90 los defectos presentes al nacimiento son cada vez más pequeños y más distales (lumbo sacros o sacros) y estamos cada vez más lejos de ver pacientes que al nacimiento se presentaban con defectos dorso lumbo sacros y cifosis.
A qué se debe este cambio? Estoy casi seguro que se debe a la aplicación en 1993 en Argentina de la ley de “las Harinas” que obligaba a los productores de Harinas de trigo y maíz a suplementarlas con ácido fólico. Estudios hechos 6 ó 7 años después demostraban que la mayor parte de mujeres embarazadas tenían llenos sus depósitos de folatos, lamentablemente también demostraron que también demostraron que la mayor parte de ellas no tenia idea que podrían haber tomado ácido fólico como complemento vitamínico, lo mismo fue observado en médicos generalistas y ginecólogos que tampoco lo indicaban. Sin embargo, que aún hoy sigan naciendo pacientes con Mielomeningocele implica que hay otros factores, algunos quizás genéticos que merecen ser investigados
El propósito de la cirugía es colocar las estructuras en una posición anatómica lo más normal posible esto es:
Liberar la médula espinal expuesta, de sus adherencias a la piel: Iniciamos la cirugía con una incisión entre el epitelio de transición y la placa medulosa, generalmente desde los costados, siendo cuidadoso cuando nos acercamos a la línea media, tanto cefálico donde se encuentra la unión del tejido nervioso malformado como caudal donde pueden estar las raíces de la cauda equina
Cerrar la médula “abierta”, de forma que tome la forma habitual (cilíndrica): Continuamos la cirugía daño puntos en los laterales de la placa medulosa, siendo conscientes de no dejar fragmentos de epitelio de transición o piel, pues de hacerlo estos más tarde darán lugar a quistes dermoides. Por otro lado deja las raíces nerviosas de la parte malformada en una posición más fisiológica disminuyendo la posibilidad de anclaje
Reponer las cubiertas: Meninges, músculo: una vez reconstruido el cilindro medular se diseca el plano dural y se recubre con puntos de seda. Luego se disecan dos laminas de fascia muscular de los músculos para vertebrales, esto nunca dejamos de hacerlo. Se han generado algunas controversias sobre si esta disección contribuye tardíamente al desarrollo de escoliosis, pero con nuestro equipo de Neuro-ortopedistas y kinesiólogos ha notado que en aquellos casos en los cuales la capa muscular es inexistente, el apoyo de una ortésis o de la mano del kinesiólogo durante las maniobras de rehabilitación genera movimientos involuntarios y dolor en el paciente. Por este motivo nosotros seguimos insistiendo en el cierre adecuado del plano muscular
Cierre adecuado de la piel por encima del defecto. Este es otro punto vital, si bien es cierto que la piel es prácticamente inexistente en esa zona, nosotros solemos disecar la piel del plano muscular para liberarla y poder cerrarla, usualmente no usamos descargas laterales si “Zetoplastias” pero siempre cuidando que la incisión principal sea vertical o sea en el sentido de la columna, permitiendo así otros abordajes como la fijación de columna si en un futuro se necesitara
Reconocimiento y exploración durante la cirugía de otras malformaciones asociadas al mielomeningocele, tales como la diastomatomyelia y diplomielia En el momento que se libera la placa medulosa y se encuentra visible el canal normal en el extremo superior del defecto, puede introducirse un elemento romo como un disector de microcirugía para explorar la presencia de espolones óseos que dividan la médula y que pueden ser reparados en el mismo momento del cierre del defecto. Para esto también es necesario contar con una radiografía de columna frente, para verificar la presencia de otras anormalidades.
Tratamiento de la cifosis Congénita: Durante mi formación en Neurocirugía este tipo de defectos era cerrado con más dificultad que los otros pero nunca planteábamos la posibilidad de realizar una corpectomía por lo sangriento de esta cirugía y la posibilidad de mortalidad intraoperatoria.
Que pienso ahora? Hemos mejorado en cuanto a las técnicas anestesiológicas y de mantenimiento intraquirúrgico de pacientes neonatos.
Es indudable que podemos cerrar mejor el defecto si no tenemos la clásica cifosis con ángulo en L1, pero hay varias cosas que todavía no podemos hacer:
No podemos estabilizar esa columna en forma definitiva, con lo cual el paciente tampoco puede ser rehabilitado prontamente, permaneciendo en decúbito dorsal por un largo tiempo. En la evolución de estos pacientes cambiamos una columna rígida en cifosis angulada en L1 por una cifosis con una curva en C más suave y más flexible pero no menos deformante e incapacitante
Estos defectos se encuentran asociados a deformidades torácicas graves algunas con fusiones costales, que son las que causan la insuficiencia respiratoria restrictiva de estos pacientes que los acompañara durante toda su vida
se asocian a Hidrocefalias y Malformaciones de Chiari, incluso hipotrofias severas de cerebelo que harán al pronóstico neurológico del paciente.
Cuando hemos ayudado a los Cirujanos Espinales en la corrección definitiva nos encontrado con sacos durales de estas cifosis nos hemos encontrado con sacos surales que contienen tejido gliótico en el cual no se identifica tejido medular sano con lo cual hemos optado por amputar el saco y reconstruirlo proximal al ángulo de la cifosis.
Quiero decir que esta corrección tardía para nada modifica el manejo que los urólogos harán del paciente, nos permite evaluar con más conciencia otros criterios como el de la suficiencia respiratoria, lo cual en este momento me lleva a pensar que la corpectomia temprana no siempre es ganancia para este grupo de pacientes
Considerando que una porción de la médula espinal se encuentra en contacto con el medio externo, y que en general hay escape de líquido céfalo raquídeo ya sea proveniente del conducto central del epéndimo, o bien del saco aracnoidal, el defecto debe ser cerrado, para evitar una infección; que en un recién nacido puede generalizarse y tener muy mal pronóstico.
Preservar la Función Neurológica
Desde ya, nosotros compartimos también la teoría dela doble lesión medular intra-útero: una por la exposición de la médula al líquido amniótico y más tarde por el roce de la placa contra las paredes del útero. Sin embargo, luego del nacimiento la exposición del tejido nervioso puede hacer que el pronóstico neurológico empeorare.
Las complicaciones pueden ser inmediatas o alejadas
Inmediatas son aquellas relacionadas al acto quirúrgico en un paciente recién nacido.
Se debe considerar aquellas relacionadas con la patología que está siendo tratada: pérdida de líquido Céfalo raquídeo a través de la herida, mala cicatrización de la herida, etc.
Según la técnica empleada, al cerrar la membrana que recubre el SNC (meninge), pueden persistir espacios entre los cuales filtra líquido céfalo raquídeo. Dicho escape, a veces queda limitado por la piel, en otros casos, termina saliendo al exterior a través de la cicatriz de la piel constituyendo una fístula.
En la zona donde se encontraba el defecto, la piel es inexistente, por lo cual algunas veces es necesario movilizar piel, que es llevada con cierta tensión, por lo que en los primeros días de postoperatorio, la herida puede tender a abrirse. Se cura entonces con el proceso conocido como “granulación”.
Alejadas, son aquellas que aparecen tiempo después de haberse realizado el cierre del defecto. Pueden estar relacionados con la aparición de sintomatología por la fijación y adherencias de la médula a la cicatriz: síndrome de médula anclada, o por la presencia de elementos de inclusión como sebo o pelos (residuos de estirpe ectodérmica de la unión piel placa medulosa) en la cicatriz del mielomeningocele: quiste dermoide
La Hidrocefalia está presente en el 95% de los pacientes con mielomeningocele. Constituye un segundo desafío para el neurocirujano, una vez que se ha realizado la plástica del defecto. Se caracteriza por la dilatación del atrio y los cuernos occipitales ventriculares (colpocefalia), configuración dada por otras malformaciones asociadas tales como: la disgenesia del cuerpo calloso, las masas intermedias (tálamos) muy cercanos entre sí
Con el advenimiento de la neuroendoscopia, la visualización de las cavidades ventriculares en estos pacientes ha aportado más datos. Por ejemplo que las masas intermedias grandes, pueden ocluir parcialmente los agujeros de Monro y que luego de colocar una derivación ventrículo peritoneal el colapso del ventrículo podría llevar a un aislamiento del ventrículo contralateral y el III ventrículo. Así mismo, permitió conocer otras alteraciones, como malformaciones estructurales del piso del III ventrículo, defectos del septum con comunicación de ambos ventrículos laterales, plexo coroideo más grande que el los pacientes sin mielomeningocele, heterotopias en las paredes ventriculares
Impedimento en la circulación del LCR a través del acueducto de Silvio el cual se encuentra estenosado
Impedimento a la circulación desde el IV ventrículo al espacio aracnoideo por malformación de los agujeros de Luschka y Magendie y el descenso de las amígdalas cerebelosas por debajo del agujero occipital
Espacio aracnoideo mas engrosado por el que el LCR circula deficientemente.
En general la hidrocefalia en el paciente con mielomeningocele, es detectada por un aumento progresivo del perímetro cefálico por encima de la curva normal, con diastásis universal de suturas, fontanela llena, ojos en sol naciente. Pocos pacientes presentan sintomatología de hipertensión endocraneana como vómitos e irritabilidad. Sin embargo, la concomitancia de hidrocefalia y alteraciones en la fosa posterior provoca en algunos casos la exacerbación de síntomas de la malformación de Arnold Chiari, tales como estridor, posición opistotónica y apneas. Mejoran al colocar el Shunt.
Hay que mencionar aquellos casos en los cuales con posterioridad al cierre del MMC se produce pérdida de LCR a través de la herida quirúrgica, o abultamiento en la herida como consecuencia de la acumulación de líquido. Estos datos bastan para indicar con seguridad el tratamiento de la hidrocefalia, por más que los estudios de Imágenes (Ecografía, TC o RMI) no muestren claramente evolutividad (aumento franco del tamaño ventricular)
En algunos pacientes, no observamos ninguno de los síntomas mencionados precedentemente, pero los estudios demuestran una ventriculomegalia. Solo la adecuada observación clínica y el seguimiento con estudios neurofisiológicos colaborarán con nosotros en la toma de decisión para colocar la derivación.
La Ecografía cerebral en el recién nacido es quizás el estudio, de elección, por la posibilidad de realizarlo sin anestesia y en la cuna del paciente, sin embargo, son la tomografía computada cerebral y la resonancia magnética de cerebro, las que nos aportarán más datos sobre la ventriculomegalia y otras malformaciones presentes en el paciente con mielomeningocele. Por otro lado, desde el punto de vista de seguimiento cobra notable importancia la realización de una tomografía computada, puesto que la ecografía cerebral es dependiente de la permeabilidad de la fontanela anterior. A lo largo del tiempo deberemos decidir por uno de los estudios teniendo en cuenta la radiación que transmite la Tomografía o la duración de la anestesia para la realización de la Resonancia
El tratamiento de elección de la hidrocefalia es la derivación ventrículo peritoneal, la que en la mayor parte de los casos es colocada dentro de los dos primeros meses de vida.
No debemos demorar tanto la decisión puesto que la macrocefalia producida implica un cráneo más grande, no un cerebro más grande con lo cual al colocar la derivación tardíamente nos exponemos a la aparición de colecciones subdurales post shunt
Otras veces el ventrículo contralateral al que tiene el catéter queda excluido, como consecuencia del colapso del ventrículo en el cuál se encuentra el catéter, se ocluye el agujero de Monro por la masa intermedia agrandada, esto es una situación vista con frecuencia en nuestros pacientes y que no requiere tratamiento
Algunos autores han propuesto últimamente como una alternativa para estos pacientes, en especial para el grupo de pacientes que presentan disfunciones valvulares repetidas (más de dos en un año), la realización de una fenestración endoscópica del III ventrículo. Esto en nuestra experiencia, es muy difícil de llevar a cabo, puesto que el 80 a 90% de los pacientes tiene anatomías ventriculares alteradas, lo que hace prácticamente imposible planear una cirugía neuroendoscópica. Por otro lado las comprobaciones anatomo-patológicas y embriológicas muestran alteraciones en el espacio aracnoideo de estos pacientes que lo vuelve incapaz de permitir una circulación normal del LCR.
En nuestro Servicio de Neurocirugía la etiología de Hidrocefalia más frecuentemente tratada es la congénita ligada al Mielomeningocele.
No obstante que la colocación de la prótesis implica un gran beneficio en la protección de la corteza cerebral, hemos de tener en cuenta, que existen complicaciones como la disfunción valvular y la infección del sistema. En nuestro consultorio Multidisciplinario casi un 32% de los pacientes ha presentado un episodio de disfunción valvular y más de la mitad de ellos más de dos episodios, esto lleva a un segundo: las infecciones por shunt en este grupo alcanzan el 17%.
En la búsqueda de estas complicaciones se deberán realizar los siguientes estudios: ecografías cerebrales, radiografías del sistema de derivación, y tomografía computada cerebral cuando lo que se sospeche sea una disfunción del sistema; y el agregado de una muestra de líquido céfalo raquídeo y una ecografía abdominal, cuando quieran descartar colecciones líquidas relacionadas con infecciones.
Disfunción del sistema de derivación
La gran mayoría se deben a oclusiones parciales del catéter ventricular, quedando el resto repartido entre desconexiones en algún punto del sistema, fallas de la válvula, catéter distal corto, y rupturas del catéter distal, que generalmente ocurren en prótesis que llevan mucho tiempo de colocadas y en las cuales se ha desarrollado una fibrosis peri-catéter, con posterior infiltración de calcio, aumento de la porosidad del material de la prótesis y finalmente ruptura. Estos pacientes en general y al menos por un tiempo toleran la disfunción con pocos síntomas, puesto que la fibrosis peri-catéter actúa manteniendo la permeabilidad y continuidad del sistema. No obstante se debe ser prudente, y el problema hay que solucionarlo ni bien hecho el diagnóstico.
Infección del sistema
Debe tenerse en cuenta que la manipulación de la prótesis durante la colocación o revisión de un sistema conlleva un riesgo importante de infección, superior al de otras neurocirugías. Sin embargo, siempre debe llevarse a cabo una cuidadosa anamnesis y examen clínico en estos pacientes, dado que la primera causa de síndrome febril es por lejos la infección urinaria. La sospecha de infección del sistema surge cuando encontramos un cuadro clínico de disfunción valvular, cercano a un procedimiento quirúrgico sobre la prótesis (se considera que la infección puede ser posible hasta una año después de un procedimiento quirúrgico sobre la válvula). Más aún si se comprueba la presencia de un pseudoquiste abdominal, que surge como una reacción del peritoneo ante un LCR infectado. En estos casos, ayuda mucho la obtención de una muestra de LCR previo al retoque del sistema.
En caso de comprobarse un LCR alterado con características de infección, el paso siguiente es la extracción del sistema de derivación, y a partir de ese momento se decidirá, el manejo de la hidrocefalia, ya sea mediante la colocación de un drenaje ventricular externo (DVE), o con la realización de punciones ventriculares, de acuerdo con la sintomatología. (Más de dos punciones ventriculares por día por síntomas francos de Hipertensión endocraneana, es indicación de colocación de DVE).
La mayor parte de las infecciones son producidas por gérmenes de la piel, (ej. estafilococo) de ahí que siendo de baja virulencia exista una “convivencia” del germen con el huésped.
Una situación que no por menos frecuente deja de ser importante, es la relacionada con las derivaciones ventrículo atriales, en las cuales la infección es detectada por la presencia de una Shunt-nefritis (en las infecciones a estafilococo coagulasa-), o en otros casos más severos por la presencia de una endocarditis bacteriana con vegetaciones en la válvulas cardíacas de las cavidades derechas.
Chiari en 1896 (Ueber Veränderungen des Kleinhirns, den pons un der medulla oblongata in folge von congenitaler Hydrocephalie des Grosshirns. Denkschirft Acad Wiss. Wien, 63, 71-116), describió una serie de alteraciones anatómicas y estructurales del cerebelo y el tronco cerebral, algunas de las cuales aparecían en los pacientes portadores de defectos del tubo neural.
En su descripción inicial Chiari, se refiere fundamentalmente a las alteraciones del cerebelo, encontrando un descenso de parte del cerebelo (amígdalas cerebelosas y vermis), más allá del agujero occipital.
Actualmente La Malformación de Chiari se refiere a un conjunto de malformaciones más complejas que involucran además del descenso amigdalino, al vermis cerebeloso, tronco cerebral y IV ventrículo, en pacientes con Mielomeningocele.
En los pacientes con Mielomeningocele existe una serie de anormalidades que involucran al cerebelo, el tronco cerebral y por ende al IV Ventrículo
Dichas anormalidades son fundamentalmente: descenso de las amígdalas cerebelosas y el vermis por debajo del agujero occipital, la unión del tronco cerebral y la médula espinal también se encuentra desplazada hacia abajo. Existe una fijación anormal entre el vermis y el tronco cerebral que dificulta la normal circulación del LCR. Hacia arriba nos encontramos con una posición baja de la tienda del cerebelo, que permite que parte del cerebelo se extienda superando los límites que le proporciona dicho repliegue meníngeo.
Esta disposición anatómica alterada a su vez tiene mucho que ver con la ocurrencia de la hidrocefalia.
El cerebelo tiene una función primordial relacionada con el equilibrio y el mantenimiento de la postura.
El tronco cerebral, regula a su vez funciones “automáticas” tales como la respiración y la frecuencia cardiaca y sirve para correlacionar funciones tales como la visión y la audición (ante un determinado ruido, dirigir la mirada hacia donde pudo haberse originado). Dentro del tronco cerebral se encuentran centros nerviosos que regulan el movimiento de las cuerdas vocales y coordinan estos movimientos con la respiración y la deglución.
Es muy importante mencionar que si bien la mayor parte de los pacientes con mielomeningocele tienen malformación de Arnold Chiari, no todos presentan síntomas atribuibles a esta.
Los síntomas que cuando aparecen, alertan sobre la presencia de esta malformación son:
Estridor
0Es un ruido producido durante la respiración, que denota dificultad en la toma de aire. En algunos casos se acompaña de una coloración azulada de los labios y bajo las uñas (cianosis) Tiene que ver con una incapacidad para movilizar algunas estructuras de la laringe (cuerdas vocales que permanecen en una posición medial) que dificultan la entrada de aire, y por ende la oxigenación de la sangre. Este síntoma puede aumentar con el llanto, que algunas veces es débil.
Apneas
es un trastorno en el cual la pausa entre una respiración y la siguiente se encuentra aumentada. Algunas veces a un punto tal que hacen disminuir la frecuencia cardiaca con el consiguiente riesgo de vida. Esto ocurre fundamentalmente durante el sueño, en oportunidades puede ser detectado cuando se refiere que el paciente interrumpe el sueño y se despierta varias veces.
Trastornos al tragar
Referidos por los padres como “ahogos” durante la alimentación fundamentalmente de líquidos, o bien como cuadros bronquiales a repetición que son el reflejo de pequeñas micro aspiraciones de alimento hacia la vía respiratoria.
Debilidad en los miembros superiores
Relacionada con la motricidad fina. En casos avanzados involucra la totalidad de la fuerza en los brazos.
Hay un grupo de pacientes que inicialmente se presentan con gran inestabilidad de tronco, poco progreso con la rehabilitación y un nistagmus lateral en los momentos en que intentan fijar la mirada, estos se corresponden con hipotrofias severas del cerebelo o lo que en la clasificación nueva de malformaciones cerebelosas se llama Malformación de Chiari tipo IV
Tomografía computada cerebral y de columna cervical: aporta datos indirectos tales como:
El ángulo formado por los bordes posteriores de los peñascos es más agudo.
Ausencia de cisterna magna.
Herniación de tejido cerebeloso a través de la tienda del cerebelo
IV ventrículo pequeño o no visible.
Resonancia magnética por imágenes de cerebro y columna cervical
Es el estudio de elección, puesto que permite conocer con exactitud el nivel del descenso del tronco cerebral y el cerebelo, así como la presencia de formaciones quísticas intramedulares o bulbares (Siringomielia y siringobulbia).(figura B)
Potenciales evocados auditivos y somato sensitivos de miembros superiores
Se trata de un estudio de los llamados neurofisiológicos, en los que lo que se trata de valorar el grado de alteración funcional existente en el tronco cerebral y la médula cervical. Se efectúa mediante la producción de un estímulo (en el caso del auditivo se trata de un sonido) el cuál es conducido a través del nervio auditivo y luego por vías intrínsecas del tronco hasta su arribo a la corteza cerebral. Valorando la demora en la conducción y la configuración de las ondas, se puede inferir el grado de compromiso causado por la malformación de Arnold Chiari.
Estudio poligráfico de sueño
Este estudio neurofisiológico se realiza durante el sueño y permite detectar episodios de apneas, la duración de las mismas, si se acompañan de trastornos cardiológicos (bradicardia) y si son obstructivos o centrales.
Sintomatología inherente a la malformación: apneas, estridor, trastornos deglutorios.
Estudios neurofisiológicos y de imágenes francamente alterados, o que muestran deterioro progresivo.
Previo al tratamiento de otras patologías tales como: escoliosis, siringomielia, corrección de malformación de la médula hendida, síndrome de médula anclada.
Cuando se encuentra indicado, nosotros realizamos una cirugía “descompresiva”, es decir una cirugía dirigida a aliviar la presión que estructuras óseas y ligamentarias de la columna cervical ejercen sobre estructuras vitales como el tronco cerebral. Para ello se realizan laminectomías cervicales, con resección de los ligamentos. Hace 20 años nosotros teníamos un tratamiento más agresivo que incluía la apertura de la duramadre con plástica de la misma con elementos autólogos (fascia muscular), posteriormente lo abandonamos por la gran cantidad de accidentes por sangrados a nivel de senos venosos en la unión bulbo medular casos es necesario ampliar el agujero occipital, para darle más lugar al cerebelo y el tronco. Debe tenerse especial cuidado en esta cirugía, puesto que en la duramadre existen lagos venosos anómalos, además de los senos venosos que se encuentran en una ubicación más baja y que, en el caso de romperse, pueden dar lugar a sangrados en algunos casos mortales.
Anteriormente, cuando se realizaba la apertura de la duramadre, se procedía, a liberar adherencias aracnoidales, y en algunos casos se resecaban las amígdalas cerebelosas y el vermis para liberar la salida del IV ventrículo. Esto implicaba en más de una oportunidad, un aumento de la morbilidad, incluso con alteraciones cardíacas (bradicardia o paro cardíaco) intra operatorias. Esto se produce por la gran cantidad de adherencias entre el cerebelo y tronco y la labilidad intrínseca de este último.
Deterioro de la función respiratoria: Sobre todo en pacientes con sintomatología grave, en los cuáles luego de la cirugía puede observarse depresión respiratoria, quizás debido al trauma quirúrgico sobre el tronco cerebral
Dolor: relacionado con el acto quirúrgico sobre los músculos paravertebrales.
Fístula de LCR: luego de la apertura y plástica de la duramadre durante la cirugía.
Alejadas
Reosificación: de las estructuras de la parte posterior de la columna cervical, volviendo a comprimir la unión bulbo-medular.
Los términos siringomielia e hydromyelia se han acuñado para un diverso número de patologías, que en definitiva representan la dilatación quística de la médula. Por lo cual en este caso hemos decidido utilizar ambos términos indistintamente para referirnos a la misma entidad.
Es notable como la mayor parte de las veces la siringomielia no se evidencia en la RMI inicial realizada al paciente con mielomeningocele. Esto se explicaría por un canal ependimario abierto a nivel de la placa medulosa, que en principio permite que todo el LCR se pierda a este nivel, pero una vez cerrado el defecto y no teniendo otra salida el LCR comienza a acumularse en el interior de la médula.
Dado que se trata de pacientes con mielomeningocele, en los que existen previamente trastornos deficitarios motores y sensitivos, muchas veces es muy difícil aseverar que un deterioro se deba a la presencia o evolutividad de una cavidad siringomiélica. Sin embargo, la presencia de una curva escoliótica en aumento, sin otro síntoma deberá hacernos sospechar la presencia de siringomielia.
Además hemos de mencionar que algunos de estos pacientes pueden sufrir exacerbación de sintomatología de la malformación de Arnold Chiari, producida por grandes cavidades medulares que afectan la médula cervical y la unión bulbo medular.
Es el estudio más importante puesto que permite detectar la presencia de esta patología, así como conocer la ubicación exacta, la extensión de la médula que abarca, y si produce una dilatación tan significativa que adelgaza y comprime la médula contra el canal dural.
Tomografía computada de columna
Alcanza para detectar la presencia de la patología Aunque no permite definir con claridad sus límites y la relación del quiste con el tejido nervioso.
En el servicio de Neurocirugía del Hospital de Niños, el tratamiento de elección es la derivación del LCR a peritoneo, o sea la derivación siringo peritoneal.
Otros tratamientos propuestos son la derivación siringo subaracnoideo, la que la mayor parte de las veces fracasa por: A) alteraciones malformativas de la aracnoides, secundarias a la falta de cierre del tubo neural, el trabeculado aracnoidal es mas grueso e impide la circulación del LCR. B) situaciones como el engrosamiento de la aracnoides secundario al cierre del mielomeningocele, C) alteraciones esqueléticas y anatómicas del SNC (descenso del tronco cerebral y cerebelo) que obran en contra de una normal circulación y reabsorción del LCR.
En algunos casos se propone la realización de una derivación siringo pleural. Nuestros resultados han sido malos dependiendo de alteraciones de la caja torácica (presencia de malformaciones vertebrales, escoliosis, cifosis, malformaciones costales), así como del nivel neurológico, que harán mas o menos tolerable la presencia del líquido en la cavidad pleural.
En la experiencia del Hospital de Niños que coincide con la de otros autores como Hoffman, cuando se está ante la presencia de una cavidad siringomiélica junto con malformación de Arnold Chiari II, preferimos en primer lugar realizar una descompresiva a nivel de la región occipito cervical, sin movilización de las amígdalas cerebelosas y posteriormente la inserción de una derivación siringo-peritoneal. El motivo de esta decisión es la posibilidad de incrementar notablemente los síntomas relacionados con la malformación de Arnold Chiari por la descompresión brusca generada por la evacuación de LCR.
Las complicaciones de dejar librada a su evolución natural a la siringomielia (no tratamiento), son en general los trastornos relacionados a la progresión de esta dilatación quística en el bulbo (siringobulbia) con compromiso de los pares craneanos y trastornos respiratorios. Por otro lado el deterioro irreversible de los valores neurológicos previos por lesión medular debido a compresión crónica.
Las complicaciones inherentes al procedimiento quirúrgico, son similares a las de la derivación para hidrocefalia, o sea Infección y disfunción, observando con muy baja frecuencia lesión neurológica por la mielotomía.
How to cite this article: Dabdoub CF, Dabdoub CB, Villavicencio R, Germán Quevedo. Como Lo Hago Yo: Mielomeningocele En Bolivia. Surg Neurol Int 10-Mar-2014;5:
In the world between 300.000 to 500.000 of children are born with NTD every year.[916] Most of them in developing nations. There is a difference in treatment criteria according to the economic status of the countries were the children are born.
For long a passive approach was accepted. Haimburger and Haimburger[7] have documented this trend. After the studies by Chambers and Hamburger[46] the wave was to favor early intervention.
Almost 30 years ago Ausman[1] pointed out that the treatment of MMCL was not a matter of surgical technique but depended of a series of cultural and social factors.[2] The quality of life of children with MMCL also has improved thus questioning the validity of opinions such as expressed by Lorber[10] or the Groningen protocol.[15]
South America occupies a surface of almost 18.000.000 km² with a population of about 390 millon people in 12 countries, two of which have a very high Human Development Index (HDI), six with high and four medium [Table 1].
Table 1
South American countries
Countries. Surface. Population. IDH Ranking. MA = very high A = high, M = medium
Cooperative Latin American Study of congenital malformations
This program studies the risk factors for developing congenital malformations in Latin America[3]. It began its activities in 1967, limited to Buenos Aires, then gradually expanded to include 10 countries and Costa Rica and the Dominican Republic. The hospital network attends at 200.000 live births per year. All the malformations diagnosed in children with a weight above 500 grams are registered according to protocol.
Between 1995-2008 there were 2,209,407 live births in the participating countries, 2.7% were malformed. Boliva had an incidence of 2.2% [Table 2]. Between 1998-2005 there was a significant reduction of anencephaly and spina bifida in Argentina and Chile. In the other countries there was an increase in cases.[13] The study has determined that 8.2 per 10.000 is the median for spina bifida in the region.[13]
Table 2
Geographical breakdown of total births, live births, stillbirths, total malformed malformed living, malformed stillbirths. Rates per 100[13]
International Tethered Cord Partnership
This is an initiative that is worth praising. It included 7 countries (Argentina, China, Guatemala, Mexico, Nicaragua, Nigeria and Panama). The objective is to determine the impact of early intervention on clinical evolution of the patients when diagnosed after birth and when presenting symptoms of tethered cord. An web centered data base was created for the paricipitants to download their cases.[ 12 ]
Folic Acid in South America
Supplementing flour with folic acid became a relity in 1990. It is considered an important factor in the reduction of spina bifida cases.[ 14 ] By supplementing wheat flour levels the access of the population to the nutrient bridgin over economical inequalities.[ 16 ]
Chile began fortifying flour in 2001 and undtil 2007 had a significan decrease (P<0.02) of close to 60% [ Table 3 ]. The data for Argentina, Brasil and Chile are summarized there.
Table 3
Birth prevalence rates of neural tube defects (isolated and total) in pre- and post-fortification of flour with folic acid in 3 South American countries periods[13]
Declaration of Santa Cruz de la Sierra
At the first Latin American Congress of Pediatric Neurosurgery that took place in Santa Cruz de la Sierra (Bolivia) representatives from Argentina, Bolivia, Brasil, Chile, Colombia, Ecuador, EL Salvador, Honduras, México, Panamá, Perú and Venezuela signed in April 29 2006 a pronouncement about neural tube defects stating that;
The high number of patients with myelomeningocele is a social problem
There is evidence about the efficacy of prevention through folic acid fortification
Recommended the governments of the region to support plans prevention before conception.
Recommended considering environmental factors that could be responsible for NTD such as soil fertilizers.
Today other countries of Latin America and the Caribbean have adhered to this plan and there are national regulations mandating the fortification of flour with micronutrients as recommended by the WHO.[ 5 ]
Seventy patients with MMCL seen by a multidisciplinary team at Hospital Universitario Japones (HUJ) between 2008 and 2011. The mothers were from a low and middle economic level.
14 (20%) without evident deficit; 23 (32.9%) partial neurological impairment and 33 (47.1%) paraplegia bellow the lesion
Mortality within 30 days: 7 (10%) (4 post op and 3 did not have surgery) Of the 70 MMC, three (4.3%) were not operated due to clinical condition (severe cardiopathy, extreme prematurity and hidranencephaly); five (7.1%) were transferred to another hospital due to lack of beds at HUJ, in 2 (2.86%), the parents requested discharge Over 60 patients that were operated
Time delay since birth till surgery: Less than 24 hours: 16 (26.7%); 24-48 hours 12 (20%); more than 48 hours: 32 (53.3%)
Closure: Five layers (arachnoid, dura, fascia, subcutaneous tissue and skin): 4 (6,7%); Four layers: 47 (78.3%) Three layers: 9 (15%)
Complications within 30 days of surgery: Wound infection or dehiscence 10 (16.6%); CNS infection: 7 (11.7%): CSF fistula 6 (10%)
Mortality within 30 days of surgery: 4 (6.7%)
Hydrocephalus within 30 days of surgery: 48 (80%).
Comparative analysis
Our data was similar that to that of La Paz. Santa Cruz is at 1370 feet above the sea level and La Paz at 11,900 feet.
The parameters compared in Table 4 are: Primiparous/Multiparous. Prenatal control. Rural/Urban. Admission at less than 24 hours. Admission older than 24 hours. Gender Male/Female. Lumbar-Sacral. Open/closed. Paraplegia. General mortality 30 days. Postoperative mortality.
Table 4
Data on two series of MMC in Bolivia
Fetal Surgery
One in 2013. At Hospital Caja Petrolera de Salud in Santa Cruz de la Sierra. Mother, 26 years. Defect, L5-S1 at 26 weeks. Delivery at 33 weeks. Excellent wound healing. At birth head circumference 35 centimeters (+SD) and normal soft spot. Lower muscle tone in lower limbs. Adequate sensory response [ Figure 1 ].
Figure 1
Clockwise. (a) 3D echography. (b) Operative field. (c) Dura patch (Gore-Tex). (d) Surgical scar at birth
Norms at HUJ
Parents are informed that the surgery will not correct the deficit. They have ample opportunities for asking questions. We explain that we choose not to treat children who have associated conditions that endanger their lives
We aim at intervening as soon as possible without risking the chance of the child developing a CNS infection that will risk his/her more important capital, IQ. This considering that little can be done regarding the already present sensory and motor deficit
In children older than 48 hours with open MMCL we perform at least 2 laboratory test of their CSF. If there are signs of infection we treat it accordingly
We first treat the MMCL and then the hydrocephalus. We shunt with a medium pressure valve donated by an NGO (Fundacion Sonrisa Feliz) because they are not covered by insurance
ATB treatment in case of meningitis
If the child does not have hydrocephalus at the moment of discharge we schedule appointments for 3, 6 and 12 months after. In our experience Hydrocephalus manifest within 1 year
If the child presents with swallowing or breathing difficulties secondary to Chiari II we intubate him/her and then treat the associated hydrocephalus. If this does not help we consider cervical laminectomy and or occipital craniectomy.
In Bolivia as in the rest of Latin America socio economical factors weight on the prevention and treatment of MMCL We do not have statistical data but it seems that in Bolivia we have a lower incidence of NTD than in Nicaragua and Guatemala where they see 10-15 cases per month in Nicaragua and 15-20 in Guatemala[12]
Preventive use of folic acid is not fully complied in Bolivia. In South America in a study of 2810 women it was observed that only 14.8% ingested vitamin supplements with folic acid during pregnancy, and only 1% did it correctly.[9]
Even though since 2001 the Seguro Materno Infantil (SUMI) covers for prenatal testing, only 39% of women take advantage of it. Lack of adequate equipment and personnel is a factor but also the program is not widely advertised in rural areas
The referral system to tertiary care hospital is deficient. The number of home deliveries is high (24.3%) but lower that in the last census (32.7%)
Due to lack of advanced imaging there is no adequate assessment of long-term consequences of MMCL such as Chiari II, syringomyelia and tethered cord
There is poor follow up of patients from rural areas
Only 9 (15%) of the patients who were operated had 3 years of follow up. MRI was requested in those who tethered cord was suspected (motor weakness, sphincter dysfunction, pain, scoliosis).[8] Tethered cord was diagnosed in 3 of the 9 children for whom surgery was performed
Parents and Neurosurgeons and the health care team, physicians, nurses, physical therapists, are fully aware about the phenomenal challenges represented by each one of the children born with Myelomeningocele.
Anualmente nacen en el mundo entre 300.000 y 500.000 niños con alguna malformación del tubo neural (MTN),[916] siendo que en los países en vías de desarrollo su incidencia aumenta dos o tres veces más, diferencia que guardaría relación con su nivel socio-económico. Del mismo modo, la evolución de estos enfermos varía notoriamente de acuerdo al país donde nacen. Entre las MTN, destaca el mielomeningocele (MMC) por su mayor frecuencia y por ser la causa más incapacitante entre todas. Según Heimburger y Heimburger,[7] la tendencia en su tratamiento ha ido cambiando. Hace poco más de medio siglo, la conducta quirúrgica era expectante hasta los 6 meses de edad, pero después de conocer los resultados de Chambers y Heimburger,[46] este enfoque fue modificándose hasta ser hoy la reparación del MMC lo más precoz posible la decisión más generalizada.
Hace casi 30 años, Ausman[1] afirmaba que el tratamiento del MMC no era simplemente una decisión técnica, que dependía de factores culturales, religiosos, niveles educativos y socioeconómicos de la sociedad, así como de otros aspectos de carácter legal, médico y ético.[2] Como estas razones continúan siendo vigentes, el tratamiento del MMC aún es controversial. Al mismo tiempo, la calidad de vida de estos niños ha mejorado notoriamente en las últimas décadas, poniendo en entredicho algunas opiniones, como las de Lorber[10] o del conocido protocolo de Groningen.[15]
América del Sur ocupa una superficie de casi 18.000.000 km² y tiene una población cercana a los 390 millones de habitantes. Incluye actualmente 12 países, dos de los cuales tienen un Índice de Desarrollo humano (IDH) muy alto, seis poseen un IDH alto y cuatro alcanzan un IDH medio [Tabla 1].
Tabla 1
Pasíes que componen América del Sur (elaboración propia)
Estudio Colaborativo Latino Americano de Malformaciones Congénitas
Es un programa de investigación clínica y epidemiológica sobre los factores de riesgo en defectos congénitos detectados en una red de hospitales de América Latina.[ 3 ] ECLAMC comenzó sus actividades en 1967, primero limitado a la ciudad de Buenos Aires, Argentina, expandiéndose gradualmente a otros 10 países de Sudamérica, incluyendo a Costa Rica y República Dominicana. La red de hospitales de maternidad de ECLAMC examina alrededor de 200.000 nacimientos por año. Todas las malformaciones, mayores y menores, que son diagnosticadas al nacer en niños con un peso de 500 gramos o más, se registran de acuerdo con un manual de procedimientos. En el período 1995-2008 hubo 2.409.407 nacimientos en los países participantes y la tasa global de malformaciones congénitas en esta muestra fue de 2,7%. En el caso de Bolivia, alcanzó al 2,2% [ Tabla 2 ]. En el período 1995-2008, hubo una reducción significativa en las tasas de anencefalia y la espina bífida en Chile y Argentina. Sin embargo, en el resto de los países, las tasas globales de malformaciones aumentaron.[ 13 ] En años recientes, el ECLAMC ha calculado en 8.2 por 10.000 la tasa media de espina bífida en la región.[ 13 ]
Tabla 2
Distribución por país del total de nacimientos, nacidos vivos, mortinatos, total de malformados, malformados vivos, malformados mortinatos. Tasas por 100.[13] Geographical breakdown of total births, live births, stillbirths, total malformed malformed living, malformed stillbirths. Rates per 100[13]
International Tethered Cord Partnership
Otra buena iniciativa ha sido la conformación del ITCP[ 12 ] que incluye a 7 países en vías de desarrollo (Argentina, China, Guatemala, México, Nicaragua, Nigeria y Panamá), que de manera conjunta trabajan, buscando al menos dos objetivos:[ 1 ] establecer una base de datos de vigilancia internacional para examinar la correlación entre el tiempo de la corrección y los resultados clínicos en niños con espina bífida y médula anclada, y[ 2 ] impulsar la colaboración entre las instituciones internacionales sobre asuntos relacionados con la neurocirugía pediátrica. Para ello, 12 hospitales elegidos en estos países incluyeron en un banco de datos a todos los pacientes con espina bífida y médula anclada entre 0 y 15 años, y que tuvieron un seguimiento médico anual, por un periodo de 5 años.
Uso del ácido fólico en Sudamérica
El enriquecimiento de la harina con ácido fólico a partir de los años 90 del siglo pasado, con el fin de hacer prevención primaria de las MTN, fue un factor muy importante para disminuir notoriamente su incidencia.[ 14 ] Por otra parte, la fortificación de la harina de trigo es una medida que favorece la equidad, al ser su consumo universal y que a diferencia de la suplementación farmacológica, no implica desigualdades según los distintos sectores sociales.[ 16 ]
En Sudamérica, un estudio reveló que las tasas de defectos de cierre del tubo neural cayeron de forma drástica. Chile comenzó a fortificar la harina en 2001, y hasta 2007, se había logrado una disminución global significativa (P < 0.02) de cerca de 60% [ Tabla 3 ]. La frecuencia de la espina bífida disminuyó en 54%, el cefalocele en 47% y la anencefalia en un 29%. Argentina comenzó a fortificar la harina de trigo en 2004, logrando una reducción global de 44%, espina bífida, 50,3% y en la anencefalia, un 59,5%. Brasil comenzó a fortificar la harina a fines de 2005 y ha logrado una disminución, aún no significativa (23,5% en espina bífida y 54,5% en el caso de la anencefalia), seguramente porque al momento de procesar estos datos, el programa sólo llevaba menos de dos años.
Tabla 3
Tasas de prevalencia al nacimiento de defectos del tubo neural (aislados y total) en los períodos pre y post fortificación de la harina con ácido fólico en 3 países sudamericanos, (ECLAMC) (13)
Declaración de Santa Cruz de la Sierra
En el I Congreso Latinoamericano de Neurocirugía Pediátrica realizado en Santa Cruz de la Sierra, representantes de Argentina, Bolivia, Brasil, Chile, Colombia, Ecuador, EL Salvador, Honduras, México, Panamá, Perú y Venezuela, firmaron el 29 de abril de 2006 un pronunciamiento con relación al problema no resuelto de la disrafia espinal, siendo que “la mayor expresión de esta malformación –la disrafia abierta o mielomeningocele–”, “determina un elevado número de pacientes minusválidos, generando graves limitantes sociales y discriminaciones de por vida.”
Al haber “suficiente evidencia científica y experiencias nacionales que demuestran que el uso preventivo preconcepcional del ácido fólico en mujeres en edad fértil reduce significativamente la incidencia de esta malformación, y que esta es una terapia de bajo costo, sin efectos colaterales y fácilmente aplicable”, esta declaración recomendó “a los distintos gobiernos y a sus autoridades de nuestra América a considerar en forma urgente su adhesión a esta campaña preventiva de la disrafia, generando el marco legal para adicionar el ácido fólico a los alimentos de uso más popular…”.
Además, esta declaración hizo “un llamado a los gobernantes a revisa sus políticas medioambientales, pues también hay evidencia de la influencia en la aparición de estas malformaciones de distintos contaminantes químicos industriales y fertilizantes, que aparecen como cofactores etiopatogénicos en zona frutícolas o de alta industrialización”. Finalmente, el Capítulo Latinoamericano de Neurocirugía Pediátrica exhortó “a los gobiernos a asumir esta responsabilidad sanitaria en beneficios de sus pueblos y de los niños que son nuestro futuro.”
Actualmente, los países firmantes de esta declaración así como otros países de América Latina y el Caribe, han dictado regulaciones nacionales para enriquecer las harinas de trigo y maíz con los micronutrientes recomendados por la OMS.[ 5 ]
Se recopilaron los datos de 70 pacientes con diagnóstico de MMC atendidos entre 2008 y 2011 por un equipo multidisciplinario (obstetra, pediatra, neurocirujano, urólogo, ortopedista, radiólogo, psicólogo y fisiatra) del Hospital Universitario Japonés (HUJ). Por tratarse de un centro sanitario de atención pública de tercer nivel, las madres eran de nivel socioeconómico y educativo bajo o medio bajo.
Los resultados fueron los siguientes:
Del área rural provenían 23 (33%) y 47 (67%) vivían en la ciudad.
Edad de las madres: Menor a 18 años: 10 (14.2%); 18 a 25 años: 30 pacientes (42.8%); 25 a 35 años: 20 pacientes (28.5%). Mayor de 35 años: 10 pacientes (14.2%).
Edad gestacional: menos de 30 semanas: 1 (1.4%); 30 a 35 semanas: 6 (8.6%); 36 a 40 semanas: 63 (90%).
Número de parto: 15 (21.4%) fueron primíparas y 45 (78.6%) eran multíparas.
Control prenatal: 27 mujeres (38.6%). Diagnóstico de disrafia espinal: 2/27 (7.4%).
Sólo 21 mujeres (30%) ingirieron complejos vitamínicos durante el parto y apenas una (1.4%) usó ácido fólico al inicio del embarazo.
Sitio del parto: hospitalario: 53 (75.7%); partos domiciliarios: 17 (24.3%).
Sexo de los pacientes: 35 varones (50%) y 35 mujeres (50%).
Edad de ingreso del MMC al HUJ: 0 a 12 horas: 16 (22.8%) (todos nacidos en el HUJ); 12 a 24 horas: 8 (11.4%); 24 a 48 horas: 6 (8.5%); mayor de 48 horas: 40 (57.1%).
Localización del MMC: Occípito-cervical: 3 casos (4.3%); Dorsal (T8 a T12): 12 casos (17.1%); Lumbar: 32 (45.7%) (L1-L2: 18); L3-L4: 14); Sacra (S2 a S4): 13 casos (18.6%);
Tipo de MMC: cerrado: 23 (32.8%); abierto: 47 (67.2%).
Cuadro clínico: 14 (20%) niños sin déficit neurológico evidente; 23 (32.9%) con daño neurológico parcial y 33 (47.1%) con paraplejia por debajo de la lesión.
Mortalidad general a los 30 días: 7 casos (10%) (4 operados y 3 no operados). De los 70 MMC, tres (4.3%) no fueron intervenidos, por presentar defectos congénitos o estado general grave (cardiopatía cianótica severa, hidranencefalia y una prematurez extrema); cinco (7.1%) fueron transferidos a otros centros, por falta de espacio en nuestro hospital y en 2 casos (2.86%), los padres pidieron alta solicitada. Sobre un total de 60 pacientes operados, observamos:
Tiempo medio de reparación del MMC desde su nacimiento hasta la cirugía: Menos de 24 horas: 16 (26.7%); de 24 a 48 horas: 12 (20%); mayor a 48 horas: 32 (53.3%).
Modalidad de cierre: En cinco planos (aracnoides, duramadre, fascia, tejido subcutáneo y piel): 4 casos (6,7%); en cuatro planos: 47 (78.3%) y en tres planos: 9 (15%).
Días de estadía en el hospital: De 1 a 7 días: 20 (28.5%); de 7 a 15 días: 20 (28.5%); de 15 a 30 días: 30 (42.8%);
Complicaciones postquirúrgicas más comunes a los 30 días: Infección o dehiscencia de la sutura: 10 casos (16.6%); infección del SNC: 7 (11.7%); fístula de líquido LCR: 6 (10%);
Mortalidad postoperatoria a los 30 días: 4 (6.7%).
Hidrocefalia a los 30 días de vida: 48 pacientes (80%)
Análisis comparativo
En aquellos parámetros que podían compararse, los resultados alcanzados en Santa Cruz de la Sierra (416 ms. sobre el nivel del mar) fueron similares a los encontrados en un hospital de La Paz, situado en la zona montañosa de Bolivia (3.640 ms. sobre el nivel del mar),[ 11 ] excepto en cuanto a control prenatal, localización más frecuente del MMC, paraplejia, mortalidad general y postoperatoria [ Tabla 4 ].
Tabla 4
Datos estadísticas de dos series de MMC en Bolivia
Cirugía fetal
En Santa Cruz de la Sierra (Hospital Caja Petrolera de Salud) se realizó por primera vez en 2013 una cirugía fetal de un MMC a cielo abierto, acompañado de una malformación de Chiari tipo II y sin hidrocefalia. La madre tenía 26 años de edad. Se realizó el cierre hermético del defecto neural (L1-L5) en la semana 26ª del embarazo. El nacimiento de la niña se produjo a las 33 semanas, sin ninguna intercurrencia. La herida quirúrgica estaba cerrada por completo, con excelente epitelización de los bordes. El PC medía 35 cms. (+2DS) y fontanela normotensa. Al examen, presentaba hipotonía de los miembros inferiores, siendo positiva la estimulación nociceptiva [ Figura 1 ].
Figura 1
(a) Diagnóstico prenatal del MMC (L1-L5) con Ecografía en 3D. (b) Imagen in situ peroperatorio. (c) Injerto con sustituto dural sintético Goretex® (politetrafluoroetileno-PTFE). (d) Cicatriz quirúrgica del recién nacido
Algunas pautas en el HUJ
Previa a la cirugía reparadora, se informa detenidamente a los padres el objetivo del procedimiento y beneficios que se esperan alcanzar; en qué consiste la operación y cómo es el postoperatorio habitual, además de los riesgos, complicaciones y secuelas posibles. Además, insistimos que la cirugía es correctora y no curativa. Una vez que los progenitores están conscientes de la situación pueden firmar el consentimiento respectivo. Esta es una medida de cumplimiento legal obligatorio.
A menos que los padres lo soliciten, recomendamos no operar en casos con graves anomalías congénitas o enfermedades que ponen en serio riesgo la vida del paciente, teniendo en cuenta que la mayoría de estos casos suelen fallecer en las primeras semanas. Esta decisión se toma con todo el staff que acompaña a estos pacientes.
Si bien el tratamiento quirúrgico del MMC es una urgencia y no una emergencia, en lo posible intentamos intervenir de manera temprana, tratando de impedir sobre todo las infecciones del SNC, que suelen ser las causas más comunes del deterioro de sus funciones cognitivas (IQ), pues los otros daños neurológicos (paraplejia, disturbios esfinterianos, etc.), casi siempre son definitivos.
En caso que el MMC abierto tenga más de 48 horas, realizamos al menos dos análisis de LCR. Si los resultados son normales, se hace la corrección del MMC. De lo contrario, se inicia antibioticoterapia, hasta verificar que el examen del LCR sea estéril. En dos niños puncionamos el saco del MMC, siendo normal el LCR en ambos casos.
Cuando hay hidrocefalia concomitante y MMC cerrado, primero atendemos la disrafia espinal. Para disminuir el riesgo de infección del shunt, alrededor del décimo día del postoperatorio, preferimos colocar una DVP, generalmente de presión media. Las válvulas son donadas por una ONG (Fundación Sonrisa Feliz), habida cuenta que no las ofrece el seguro materno-infantil (SUMI).
En presencia de meningitis o sepsis e hidrocefalia concomitante, se inicia el tratamiento con los antibióticos recomendados. Una vez que el LCR es estéril (demostrado por cultivos seriados) se coloca el shunt.
Si el paciente con MMC no presenta hidrocefalia al momento del alta hospitalaria, el niño retorna al mes, 3, 6 y 12 meses para su control médico, porque en nuestra experiencia, la hidrocefalia suele manifestarse generalmente antes del año de edad.
Cuando la malformación de Chiari asociada al MMC presenta disfunción de la deglución y amenaza con obstrucción de las vías aéreas, se considera la intubación endotraqueal. Si hay una hidrocefalia asociada, se trata primero ésta, ya que con el shunt suele mejorar el cuadro clínico. De lo contrario, puede recurrirse a una cirugía descompresiva a nivel de fosa posterior con laminectomía cervical alta, según sea el caso.
En Bolivia, igual que otros países sudamericanos con IDH medio, un mayor porcentaje de familias con nivel socio-económico y cultural bajo tienen hijos con MMC, lo que apoya la hipótesis que factores nutricionales y la falta de prevención, juegan un rol importante en el desarrollo de la MTN. Aunque no contamos con datos estadísticos confiables, parece que nuestra incidencia es menor que en otros países latinoamericanos. Por ejemplo, estudios realizados en Nicaragua y Guatemala, llegan a reportar mensualmente entre 10-15 enfermos y 15-20 casos de espina bífida, respectivamente.[12]
El uso preventivo del ácido fólico en edad fértil no se cumple en Bolivia como lo establece una norma legal desde 1996. Faltan más control y difusión en la población sobre las ventajas que ofrece la fortificación de la harina y cereales con ácido fólico. Un amplio estudio realizado en 30 hospitales de Sudamérica (2810 mujeres postparto), demostró que sólo 14.8% de mujeres habían ingerido suplemento vitamínicos conteniendo ácido fólico para evitar una MTN, mientras que apenas un 1% habían cumplido de manera adecuada su prevención,[9] cifra que coincide con nuestros hallazgos.
Pese a contar desde 2001 con el SUMI, que permite a la mujer embarazada realizar controles prenatales cada mes, observamos en nuestra serie que apenas un 39% lo realizó, demostrándose que se desconoce este programa social, sobre todo en el área rural y zona periurbana. Además, muchos de los centros de salud no cuentan con el equipamiento apropiado y profesionales capacitados, para realizar el diagnóstico prenatal de estas malformaciones.
Aún no funciona adecuadamente el sistema de referencia y contra-referencia que permita trasladar al paciente de manera oportuna a centros de tercer nivel, razón por la cual mucho de los pacientes con MMC son atendidos después de las 48 horas de vida. Asimismo, los partos domiciliarios todavía muestran una alta incidencia (24.3%); sin embargo, este porcentaje es inferior a la registrada en el último Censo nacional de 2012 (32.7%).
Debido a la falta de acceso en la mayoría de los casos a resonancia magnética nuclear (RMN) y/o tomografía axial computadorizada (TAC), no se puede valorar adecuadamente algunas complicaciones, generalmente añadidas al MMC (malformación de Chiari, médula anclada, siringomielia, etc.).
Hay dificultad en el acompañamiento adecuado a estos enfermos, sobre todo quienes viven en el área rural, lo que impide realizar sus controles, así como el tratamiento con otras especialidades médicas y su rehabilitación.
Sólo 9 (15%) de los MMC intervenidos tuvieron por lo menos 3 años de seguimiento. Se indicó RMN en quienes presentaban sospecha de médula anclada (mayor debilidad muscular, empeoramiento de la marcha, disturbios esfinterianos, dolor, deformidades ortopédicas o escoliosis),[8] demostrándose esta patología en 3/9 (33.3%), por lo que fueron operados.
La sociedad civil, los padres de hijos con MMC, los neurocirujanos y el equipo profesional que atienden a estos pacientes, saben que un recién nacido con esta malformación es un gran desafío para el sistema de salud de cualquier país.[9]
6. Heimburger RF. Early repair of myelomeningocele (spina bifida cystica). J Neurosurg. 1972. 37: 594-600
7. Heimburger RF, Heimburger DC. Reflections on a career in neurosurgery. Surg Neurol Int. 2013. 4: 89-
8. Hudgins RJ, Gilreath CL. Tethered spinal cord following repair of myelomeningocele. Neurosurg Focus. 2004. 16: 1-4
9. Lazareff JA.editors. Neural Tube Defects. Myelomeningocele. World Scientific Publishing, Co. Pte. Ltd; 2011. p. 29-91
10. Lorber J. Results of treatment of myelomeningocele. An analysis of 524 unselected cases, with special reference to possible selection for treatment. Develop Med Child Neurol. 1971. 13: 201-204
11. Ludueña MP, Mazzi Gonzales de Prada E. Clinic characteristics of myelomeningocele in newborns admitted to the Hospital del Niño “Dr. Ovidio Aliaga Uría” 1993 2002 [Article in Spanish]. Rev Bol Ped. 2003. 42: 160-165
12. Mulholland CB, Aranda G, Arredondo LA, Calgua E, Contreras F, Espinoza DM. The International Tethered Cord Partnership: Beginnings, process, and status. Surg Neurol Int. 2011. 2: 38-
13. Nazer HJ, Cifuentes OL. Congenital malformations in Latin America in the period 1995-2008. [Article in Spanish]. Rev Med Chil. 2011. 139: 72-8
14. . U.S. Public Health Service: Recommendation for the use of folic acid to reduce the number of cases of spina bifida and other neural tube defects. MMWR. 1992. 41: 1-7
15. Verhagen E, Sauer P. The Groningen Protocol — Euthanasia in Severely Ill Newborns. N Engl J Med. 2005. 352: 959-962
16. Zabala R, Waisman I, Corelli M, Tobler B. Folic acid for neural tube defects prevention: consumption and information in fertil-age women in Centro Cuyo Region. [Article in Spanish]. Arch Argent Pediatr. 2008. 106: 295-301
Department of Neurosurgery, Gold Coast University Hospital, 1 Hospital Boulevard, Southport, Queensland 4125, Australia
Department of Neurosurgery, Royal Adelaide Hospital, North Terrace, Adelaide, South Australia 5000, Australia
Correspondence Address: Thorbjorn Loch-Wilkinson Department of Neurosurgery, Royal Adelaide Hospital, North Terrace, Adelaide, South Australia 5000, Australia
How to cite this article: Loch-Wilkinson T, Tsimiklis C, Santoreneos S. Trigeminal neuralgia associated with Chiari 1 malformation: symptom resolution following craniocervical decompression and duroplasty: Case report and review of the literature. Surg Neurol Int 23-Jul-2015;6:
Background:Trigeminal neuralgia (TN) may rarely be the presenting or only symptom of Chiari 1 malformation (CM). Isolated case reports have described resolution of TN following craniocervical decompression where TN is present in association with CM.
Case Report:This report discusses an unusual case of pure TN associated with CM that was successfully treated with craniocervical decompression and duroplasty and reviews the limited literature on the subject.
Conclusion:TN may be the sole presenting symptom of CM and can be successfully managed with craniocervical decompression. Clinicians should be aware of the association of TN with CM and consider surgical management.
Trigeminal neuralgia (TN) may rarely be the presenting or only symptom of Chiari 1 malformation (CM). Isolated case reports have described resolution of TN following craniocervical decompression where TN is present in association with CM. This report discusses an unusual case of this nature that was successfully treated with craniocervical decompression and duroplasty and reviews the limited literature on the subject.
A 39-year-old female presented with severe right sided facial pain of sudden onset. Her pain was so severe that she was admitted to a rural hospital for pain management. Her facial pain did not respond to oral medications including high dose carbamazepine, pregabalin, nonsteroidal antiinflammatory drugs (NSAIDS) and opiates. The patient was transferred to a tertiary hospital and investigation with magnetic resonance imaging (MRI) demonstrated a CM with tonsillar descent to the C1 level and very mildly increased T2 signal in the upper cervical cord [Figure 1]. There was no evidence of vascular conflict.
Figure 1
Preoperative T2WI MRI Brain demonstrating Chiari 1 malformation with subtle upper cervical cord signal change. The patient had severe and refractory right sided trigeminal neuralgia as the sole presenting symptom
The patient proceeded to have a craniocervical decompression and duroplasty 8 weeks after first presentation of symptoms. At surgery tight arachnoid bands constricting the cerebellar tonsils were noted and the posterior aspect of the medulla had a gliotic and tented appearance consistent with chronic compression. Division of the arachnoid bands was performed and diathermy of the cerebellar tonsils without subpial aspiration. Native pericranium was used for duraplasty. There were no surgical complications. Following surgery the patient had immediate relief of facial pain and remains asymptomatic 1 year postsurgery with a satisfactory MRI appearance [Figure 2].
Figure 2
T2WI MRI scan one year postcraniocervical decompression and duroplasty. The patient remains asymptomatic
TN associated with CM is limited to a very small number of case reports. A 2008 case report with a literature review found only 19 cases in the English language literature,[8] although in correspondence following this review several authors describe additional unpublished cases. A small number of cases in the non-English language literature can also be found.[14714] Pure TN is particularly uncommon as a presentation of CM and is limited to isolated case reports.[2791013] Presentations of TN with CM may include bilateral symptoms or be secondary to hydrocephalus.[5111214]
Postulated mechanisms of generation of TN due to Chiari malformation include (i) vascular compression at the nerve root entry zone, which could be affected by hydrocephalus or anatomic factors related to the Chiari malformation, such as a small posterior fossa; (ii) demyelination; (iii) microischaemic changes; and (iv) direct brainstem compression.[8] The spinal tract of the trigeminal nucleus has been implicated due to its dorsal location and poor myelination which potentially renders it vulnerable.[612]
Case reports describe a range of successful treatments in cases of TN associated with Chiari malformation including medical treatment with carbamazepine[7] and craniocervical decompression[31012] as with this case. A bilateral TN case with associated CM was successfully treated with bilateral microvascular decompression (MVD),[15] however, it is argued by some authors that retrosigmoid craniotomy for MVD may improve symptoms by indirectly decompressing the foramen magnum.[6] Cases with associated hydrocephalus have been treated successfully with endoscopic third ventriculostomy[11] and ventricular shunt procedures.[5] Than et al., in 2011,[12] reviewed treatments for the 20 known cases described in the English literature at that time and found 15 of the 20 patients had been treated with craniocervical decompression with a reported 73% resolution of pain symptoms.
This case report contributes to the very limited number of case reports of Chiari malformation with pure TN, and those successfully treated with craniocervical decompression and duroplasty. Neurologists and neurosurgeons should consider this diagnosis in presentations of TN with concurrent CM and consider surgical treatment.
1. Ayuso-Peralta L, Jiménez-Jiménez FJ, Tejeiro J, Zurdo M, Cabrera-Valdivia F, García-Albea E. Trigeminal neuralgia associated with Arnold Chiari malformation. Rev Neurol. 1999. 29: 1345-
2. Caranci G, Mercurio A, Altieri M, Di Piero V. Trigeminal neuralgia as the sole manifestation of an Arnold-Chiari type I malformation: Case report. Headache. 2008. 48: 625-7
3. Chakraborty A, Bavetta S, Leach J, Kitchen N. Trigeminal neuralgia presenting as Chiari I malformation. Minim Invasive Neurosurg. 2003. 46: 47-9
4. Gelabert González M. Trigeminal neuralgia as the first symptom of Chiari malformation. Neurologia. 2001. 16: 189-90
5. Gnanalingham K, Joshi SM, Lopez B, Ellamushi H, Hamlyn P. Trigeminal neuralgia secondary to Chiari's malformation-treatment with ventriculoperitoneal shunt. Surg Neurol. 2005. 63: 586-8
6. González-Bonet LG, Piquer J. Trigeminal Neuralgia: A Symptom of Chiari I. Neurosurgery. 2012. 71: 911-2
7. Iváñez V, Moreno M. Trigeminal neuralgia in children as the only manifestation of Chiari I malformation. Rev Neurol. 1999. 28: 485-7
8. Papanastassiou AM, Schwartz RB, Friedlander RM. Chiari I malformation as a cause of trigeminal neuralgia: Case report. Neurosurgery. 2008. 63: E614-5
9. Peñarrocha M, Okeson JP, Peñarrocha MS, Angeles Cervello M. Orofacial pain as the sole manifestation of syringobulbia-syringomyelia associated with Arnold-Chiari malformation. J Orofac Pain. 2001. 15: 170-3
10. Rosetti P, Oulad Ben Taib N, Brotchi J, De Witte O. Arnold Chiari Type I malformation presenting as a trigeminal neuralgia: Case report. Neurosurgery. 1999. 44: 1122-4
11. Teo C, Nakaji P, Serisier D, Coughlan M. Resolution of trigeminal neuralgia following third ventriculostomy for hydrocephalus associated with chiari I malformation: Case report. Minim Invasive Neurosurg. 2005. 48: 302-5
12. Than KD, Sharifpour M, Wang AC, Thompson BG, Pandey AS. Chiari I malformation manifesting as bilateral trigeminal neuralgia: Case report and review of the literature. J Neurol Neurosurg Psychiatry. 2011. 82: 1058-9
13. Tortosa A, Arbizu T, Ferran E, Peres Serra J. Arnold Chiari malformation presenting as trigeminal neuralgia. Neurologia. 1991. 6: 148-50
14. Uldry PA, Fankhauser H, de Tribolet N. Trigeminal involvement and peripheral facial paralysis caused by Arnold-Chiari malformation with hydrocephalus. Neurochirurgie. 1985. 31: 73-5
15. Vince GH, Bendszus M, Westermaier T, Solymosi L, Ernestus RI, Matthies C. Bilateral trigeminal neuralgia associated with Chiari's type I malformation. Br J Neurosurg. 2010. 24: 474-6
How to cite this article: Kim J, Roberts DW, Bauer DF. CSF hydrothorax without intrathoracic catheter migration in children with ventriculoperitoneal shunt. Surg Neurol Int 23-Jul-2015;6:
Background:Thoracic complications of ventriculoperitoneal (VP) shunts have been extensively reported in the literature. Cerebrospinal fluid (CSF) hydrothorax without catheter migration, however, has been rarely described and poorly understood.
Case Description:We describe development of pleural effusion and respiratory distress in a 3-year-old boy with no evidence of VP shunt catheter displacement on plain radiograph and stable ventricle size on rapid sequence magnetic resonance imaging (MRI) brain. Chest X-ray revealed complete opacity of right hemithorax. Pleural effusion was consistent with transudate. Beta-2 transferrin returned positive. The patient underwent externalization of VP shunt, and upon resolution of effusion, re-internalization with new distal shunt catheter. A literature review of CSF hydrothorax in children without intrathoracic shunt migration was performed. Eleven cases were identified in the English literature. Age at VP shunt placement ranged from birth to 8 years of age. Interval from VP shunt placement to CSF hydrothorax ranged from 1.5 months to 5 years. History of shunt revision was reported in two cases. Presenting symptoms also included ascites and inguinal hernia or hydrocele. Reported diagnostic studies consist of CSF culture, radionuclide shuntogram, beta-2 transferrin, and beta-trace protein. Laterality of the VP shunt and development of pleural effusion were predominantly right sided. Definitive surgical treatment included VA shunt, repositioning of the peritoneal catheter, and endoscopic choroid plexus coagulation.
Conclusion:CSF hydrothorax is a rare thoracic complication of VP shunt placement with no radiographic evidence of shunt migration or malfunction. Postulated mechanisms include limited peritoneal capacity to resorb CSF in children and microscopic communications present in congenital diaphragmatic hiatuses.
Complications of ventriculoperitoneal (VP) shunt have been reported extensively in the literature. Thoracic manifestations include pleural effusion, bronchial perforation, pneumothorax, and pneumonia. Cerebrospinal fluid (CSF) pleural effusion in the absence of migration of distal VP shunt catheter in children, however, has been rarely described and poorly understood.[11] We report a case of CSF hydrothorax in a child with no radiographic evidence of VP shunt migration and review the literature on associated clinical findings.
The patient is a 3-year-old boy born at 40 weeks by Cesarean section who initially presented with congenital hydrocephalus. He had right VP shunt placement at birth, with subsequent revisions at 13 and 23 months of age. He became symptomatic 10 days prior to presentation with progressive viral-like upper respiratory symptoms, including poor oral intake, intermittent fever, cough, irritability, and an ill appearance. Patient's co-morbidities include intractable epilepsy and craniosynostosis.
On examination, he was somnolent, but he would open his eyes spontaneously. He had full strength in all extremities. His shunt incisions and shunt track were nonerythematous and nontender. No swelling was seen along the track. He demonstrated intermittent cough with mild desaturations to 89%. Laboratory studies were within normal limits aside from mild thrombocytopenia (79 × 103/mcL) and elevated valproic acid level (183 mg/L; reference therapeutic range 50–100 mg/L).
Imaging
Chest radiograph revealed complete opacification of the right hemithorax with mediastinal displacement [ Figure 1 ]. Radiographic shunt series revealed radiographically intact VP shunt without migration into the thorax [ Figure 2 ]. Quick-brain magnetic resonance imaging (MRI) revealed stable appearance of the ventricles without change in ventricular size or new extra-axial fluid collections. Ultrasound of the abdomen and the thorax confirmed right sided pleural effusion, underlying atelectatic lung, and a small amount of ascites.
Figure 1
Chest radiograph demonstrates pleural effusion in right hemithorax
Figure 2
X-ray shunt series show no evidence of shunt disconnect
Hospital course
A chest tube was inserted on the right side with drainage of 800 ml of straw colored fluid under pressure. The fluid profile was that of a transudate without infection. The patient's mental status immediately improved. Chest tube output was brisk with 500 ml over the initial 24 h, and increasing to an output of approximately 50 ml/h. The valproic acid dose was decreased given the supratherapeutic level.
Beta-2 transferrin was sent from the chest tube drainage, resulting in a positive study. The final CSF culture was negative for infection. A decision was made to externalize the VP shunt at the abdomen, following which the chest tube output decreased considerably. The chest tube was placed to water seal with no re-accumulation of pleural fluid. The patient returned to the operating room for replacement and internalization of distal peritoneal tubing.
Postoperatively, the chest tube was maintained on water seal for 48 h without an increase in output. Serial ultrasound exam demonstrated no re-accumulation of pleural fluid, and the chest tube was subsequently removed. At 1 year follow-up, he has no re-accumulation of pleural fluid, and no signs or symptoms of shunt malfunction.
Upon initial presentation, the right chest opacity on chest radiograph was initially thought to be related to valproate toxicity resulting in an eosinophilic effusion. Consistent with this diagnosis was the patient's depressed mental state and thrombocytopenia. When the chest tube was inserted, however, pleural fluid analysis was negative for eosinophils. Pleural fluid was transudative with mostly macrophage predominant cellularity and no evidence of malignant cells. The patient did not have symptoms of common etiologies for transudative effusions, such as congestive heart failure (no cardiomegaly on chest X-ray, no S3 on auscultation, no hepatomegaly, normal pulses and blood pressure), nephrotic syndrome (normal urine output, normal serum albumin, no peripheral edema), and liver failure (normal hepatic function tests, normal coagulation tests). Despite improved respiratory status following pleural drainage, the patient continued to have persistent clear fluid drainage from his chest tube at approximately 50 ml/h. Beta-2 transferrin was sent, and its positive result confirmed the diagnosis of CSF hydrothorax.
Thoracic complications of VP shunts have been previously outlined into three categories:[11] intrathoracic trauma during placement of a shunt, migration of the peritoneal catheter into the chest, or pleural effusion accompanying CSF ascites. In the absence of iatrogenic injury or migration of the peritoneal catheter, symptomatic CSF hydrothorax may infrequently result with and without concomitant CSF ascites.[12] Absence of radiographic signs of shunt malfunction, disconnect, or intrathoracic migration of the catheter, however, raises a diagnostic challenge. Radionuclide shuntogram and beta-2 transferrin assays of pleural fluid are of significant diagnostic utility in suspected cases of CSF hydrothorax.
The mechanism of CSF hydrothorax in children without VP shunt catheter displacement remains less clear. Migration of CSF from the peritoneal to the pleural cavity depends on two factors: Malabsorption of CSF in the peritoneal cavity and open communication between the peritoneal and the pleural cavities to enable intraperitoneal CSF to pass into the pleural cavity.[6] A theoretical risk factor is the limited peritoneal capacity to resorb CSF in children, resulting in CSF ascites and associated pleural effusion. Possible contributory factors also include history of abdominal infection, abdominal surgery, and formation of pseudocysts.[5] Another possibility is mechanical leakage of CSF from the shunt valve, the catheter, or between their connection, which is most likely suspected to be the case in our patient.
Conduits for intrathoracic catheter migration traditionally have been suggested to involve congenital diaphragmatic hiatuses, such as the anterior foramen of Morgagni and the posterior foramen of Bochdalek.[7] In children, these areas in the diaphragm are also easily eroded or can harbor microscopic communications undetectable by thoracoscopy or nuclear imaging studies.[9] Chronic inflammation can further facilitate transudation of CSF fluid via capillary and lymphatic channels in the diaphragm. A cyclic pressure gradient, created by negative intrathoracic pressure during inspiration and positive abdominal pressure during expiration, is presumed to allow unidirectional flow of CSF.[3]
In view of these mechanistic factors, a literature review of CSF hydrothorax in children without intrathoracic catheter migration was performed. A total of 11 pediatric cases of CSF hydrothorax without intrathoracic catheter migration were identified in the English literature [Table 1]. Age at VP shunt placement ranged from birth to 8 years. Interval time to CSF hydrothorax ranged from 1.5 months to 5 years. Clinical risk factors for poor abdominal re-absorption of CSF were identified in two cases (18%), including one patient who had a prior abdominal surgery (Nissen fundoplication)[3] and another who previously had been diagnosed with necrotizing enterocolitis.[4] Interestingly, both resulted in shunt revision. By contrast, our patient underwent two prior shunt revisions in the absence of apparent abdominal risk factors.
Table 1
Series of CSF hydrothorax in children without intrathoracic catheter migration
Common presenting symptoms included ascites (N = 4) and inguinal hernia or hydrocele (N = 3). None of the patients with these symptoms had a history of shunt revision. It is widely accepted that very young children with poor abdominal re-absorptive capacity are susceptible to development of hydrocele or inguinal hernia following placement of VP shunt. While relatively little is known whether these presenting abdominal symptoms have any relation to CSF hydrothorax development, ascites appeared to occur mutually independent from inguinal hernia or hydrocele [Table 1]. Consistent with this trend, findings in our patient included mild ascites but no inguinal hernia or hydrocele.
As reported in Table 1, diagnostic studies reported in the literature, in descending order of frequency, consisted of CSF culture, radionuclide shuntogram, beta-2 transferrin, and beta-trace protein. CSF culture was documented in eight cases (73%). Radionuclide shunt study was obtained in five cases (45%). Beta-2 transferrin was sent in four cases (36%), all of which returned positive. One report utilized beta-trace protein, which was positive. Variability in diagnostic methods may be related to diagnostic preference or institutional availability of these tests as reflected by the wide geographic origin of the report series.[1]
A chest tube was placed in 5 of 11 cases (45%) for management of pleural effusion. Laterality of VP shunt was right-sided (82%), left-sided (9%), and not reported (9%). Pleural effusion developed in the right lung (82%), left lung (9%), and bilateral lungs (9%). While most VP shunts and resultant hydrothorax were right-sided, laterality of VP shunt did not overlap with concomitant hydrothorax when it occurred in the left.[2] There was one case of right-sided VP shunt resulting in bilateral involvement of the lungs.[4]
Final surgical treatment modalities ranged from ventriculo-atrial (VA) shunt placement (73%), reposition of peritoneal catheter (18%), and endoscopic choroid plexus coagulation (9%). Of eight patients in whom VA shunt was eventually placed, the catheter was externalized prior to VA shunt placement in two cases, and the peritoneal catheter was repositioned prior to VA shunt placement in one case. Externalization of a distal catheter may guide subsequent strategy for treatment, whether it is conversion to a VA shunt or replacement of the distal VP shunt catheter. Ventriculo-pleural shunt was attempted in one patient but subsequently had to be revised to a VA shunt. Further follow-up data may be useful for determination of long-term efficacy of various surgical revision modalities.
CSF hydrothorax is a rare but important complication of VP shunt placement in children without evidence of shunt migration or malfunction. Postulated mechanisms include limited peritoneal capacity to resorb CSF in children and microscopic communications present in congenital diaphragmatic hiatuses. In suspected cases, beta-2 transferrin assay and radionuclide tracer shunt series are useful diagnostic studies. CSF culture is commonly obtained to exclude infection. Thoracocentesis of pleural fluid or chest tube placement facilitates management of persistent pleural effusion. Externalization of the distal shunt catheter may resolve the hydrothorax and may guide subsequent treatment strategy.
1. Adeolu AA, Komolafe EO, Abiodun AA, Adetiloye VA. Symptomatic pleural effusion without intrathoracic migration of ventriculoperitoneal shunt catheter. Childs Nerv Syst. 2006. 22: 186-8
2. Born M, Reichling S, Schirrmeister J. Pleural effusion: Beta-trace protein in diagnosing ventriculoperitoneal shunt complications. J Child Neurol. 2008. 23: 810-2
3. Chuen-im P, Smyth MD, Segura B, Ferkol T, Rivera-Spoljaric K. Recurrent pleural effusion without intrathoracic migration of ventriculoperitoneal shunt catheter: A case report. Pediatr Pulmonol. 2012. 47: 91-5
4. Faillace WJ, Garrison RD. Hydrothorax after ventriculoperitoneal shunt placement in a premature infant: An iatrogenic postoperative complication. Case report. J Neurosurg. 1998. 88: 594-7
6. Hadzikaric N, Nasser M, Mashani A, Ammar A. CSF hydrothorax-VP shunt complication without displacement of a peritoneal catheter. Childs Nerv Syst. 2002. 18: 179-82
7. Kocaogullar Y, Guney O, Kaya B, Erdi F. CSF hydrothorax after ventriculoperitoneal shunt without catheter migration: A case report. Neurol Sci. 2011. 32: 949-52
8. O’Halloran PJ, Kaliaperumal C, Caird J. Chemotherapy-induced cerebrospinal fluid malabsorption in a shunted child: Case report and review of the literature. BMJ Case Rep. 2013. 2013: pii: bcr2012008255-
9. Patel AP, Dorantes-Argandar A, Raja AI. Cerebrospinal fluid hydrothorax without ventriculoperitoneal shunt migration in an infant. Pediatr Neurosurg. 2011. 47: 74-7
10. Smith JC, Cohen E. Beta-2-transferrin to detect cerebrospinal fluid pleural effusion: A case report. J Med Case Rep. 2009. 3: 6495-
11. Taub E, Lavyne MH. Thoracic complications of ventriculoperitoneal shunts: Case report and review of the literature. Neurosurgery. 1994. 34: 181-3
12. Ulus A, Kuruoglu E, Ozdemir SM, Yapici O, Sensoy G, Yarar E. CSF hydrothorax: Neither migration of peritoneal catheter into the chest nor ascites. Case report and review of the literature. Childs Nerv Syst. 2012. 28: 1843-8
How to cite this article: Singh M, Rios Diaz AJ, Golby AJ, Caterson EJ. “Countersinking” of reservoir in an irradiated patients can decrease tension on scalp closure. Surg Neurol Int 23-Jul-2015;6:
Background:Subcutaneous reservoirs are used to provide therapy by establishing access to cerebrospinal fluid. However, it is associated with complications such as hemorrhage, infection, malfunction, and malpositioning. In an irradiated field with thin skin, use of reservoir can result in wound dehiscence, wound infection, and device extrusion.
Case Description:We introduced a âcountersinkingâ technique for the reservoir placement which involves the creation of bony recess in the skull to effectively accommodate the reservoir and decrease the protrusion. âCountersinkingâ of the reservoir can result in tension-free closure of the scalp and allow durable coverage of the reservoir. In the representative case, the incisional wound healed completely without any concern for wound dehiscence and the countersink technique may have contributed to effective healing of the radiated scalp.
Conclusion:Countersinking of the reservoir can be a strategy to prevent complications such as wound dehiscence, and device extrusion in any patient, but in irradiated patients with very thin skin it also enables tension-free closure of the wound.
Subcutaneous reservoirs such as ommaya reservoir are used to administer intraventricular antibiotics for chronic meningitis, intrathecal chemotherapy for central nervous system lymphoma, and aspirate fluids from cystic tumors.[25101314] However, their use is associated with a complications such as hemorrhage, malfunction, and misplacement.[37891112] Placement of a protruding device underneath a thin irradiated skin can exert increased tension on the already weakened wound and result in wound dehiscence and wound infection.[146] We propose a “countersinking” technique of reservoir placement which creates a tailored bony recess to accommodate the reservoir [Figure 1].
Figure 1
Protrusion of the reservoir increases the tension on the overlying wound (a) while countersinking of the reservoir between the outer and inner table of the skin bone (b) promotes tension-free wound closure. This technique facilitates wound healing prevents complications such as wound dehiscence
The patient was a 46-year-old woman who underwent right frontal craniotomy in1998 for tumor resection. Pathology was consistent with astrocytoma requiring adjuvant chemoradiation therapy. Her follow-up magnetic resonance imaging in 2012 showed interval cyst enlargement in the right frontal lobe. A plan was made to proceed with aspiration of the cyst contents and then leave a catheter into the cavity which would then be connected to a reservoir for access in case the cyst reaccumulates.
Due to the history of radiation and very thin skin, there was a significant concern for wound breakdown and a modified technique was used for reservoir placement. A U-shaped incision was made adjacent to the previous incision outside the borders of the radiated skin and a pericranial flap was then raised. The skull bone was drilled through the outer table and the diploic layer to the level of the inner table to provide a tailored bony recess for countersinking the reservoir in the skull so that it would not protrude. The BrainLab navigation system was used to establish the oblique trajectory of the catheter. The dura was opened and the catheter was passed into the cyst. About 20 ml of yellowish thick fluid was aspirated and sent to cytology. The catheter was then secured to the reservoir which was effectively countersunk in the bony recess. The patient had no complications from the reservoir placement at 2 months follow-up appointment.
Subcutaneous reservoirs provide an effective way of establishing external access to cerebrospinal fluid (CSF) and other intracranial fluid spaces.[25] However, technical complications in the form of malpositioning and infectious complications leading to meningitis can be life threatening.[7] Placement of a reservoir in an irradiated wound with thin skin increases the risk of wound dehiscence and device extrusion. With wound dehiscence, a superficial wound infection can easily track to the CSF and intracranial cavity resulting in serious intracranial complications. Any technical modification toward preventing such potential complications would have far reaching consequences.
In our patient, there was a high risk of wound dehiscence and reservoir extrusion given the thin irradiated skin. Countersinking of the reservoir into the bone decreases the protuberance and by doing so, minimizes the stretching of the overlying skin [Figure 2]. This simple modification decreases the risk of wound dehiscence and device extrusion. It also results in effective “soft tissue lengthening” and allows a tension free closure. We used an oblique trajectory, facilitated by neuronavigation and a tracked stylet for catheter placement, to ensure that the incision is outside the boundaries of previous radiation and the reservoir is not directly underneath the incision.
Figure 2
Coronal (a) and axial (b) section of computed tomography scan demonstrating the placement of the ommayma reservoir in the bony recess of the skull through the outer table and the diploic layer up to the inner table
Since a large number of brain tumor patients require chemoradiation and these patients often have significant other co-morbidities resulting in poor wound healing, the “countersinking” of the reservoir can potentially prevent the risk wound dehiscence and device extrusion in these patients and enable tension free intra-operative closure of the wound.
1. Barnett GC, West CM, Dunning AM, Elliott RM, Coles CE, Pharoah PD. Normal tissue reactions to radiotherapy: Towards tailoring treatment dose by genotype. Nat Rev Cancer. 2009. 9: 134-42
2. Bernardi RJ, Bomgaars L, Fox E, Balis FM, Egorin MJ, Lagattuta TF. Phase I clinical trial of intrathecal gemcitabine in patients with neoplastic meningitis. Cancer Chemother Pharmacol. 2008. 62: 355-61
3. Dinndorf PA, Bleyer WA. Management of infectious complications of intraventricular reservoirs in cancer patients: Low incidence and successful treatment without reservoir removal. Cancer Drug Deliv. 1987. 4: 105-17
4. Hom DB, Adams GL, Monyak D. Irradiated soft tissue and its management. Otolaryngol Clin North Am. 1995. 28: 1003-19
5. Jiang PF, Yu HM, Zhou BL, Gao F, Shen SX, Xia ZZ. The role of an Ommaya reservoir in the management of children with cryptococcal meningitis. Clin Neurol Neurosurg. 2010. 112: 157-9
6. Kulkarni S, Ghosh SP, Hauer-Jensen M, Kumar KS. Hematological targets of radiation damage. Curr Drug Targets. 2010. 11: 1375-85
7. Lishner M, Perrin RG, Feld R, Messner HA, Tuffnell PG, Elhakim T. Complications associated with Ommaya reservoirs in patients with cancer. The Princess Margaret Hospital experience and a review of the literature. Arch Intern Med. 1990. 150: 173-6
8. Obbens EA, Leavens ME, Beal JW, Lee YY. Ommaya reservoirs in 387 cancer patients: A 15-year experience. Neurology. 1985. 35: 1274-8
9. Ratcheson RA, Ommaya AK. Experience with the subcutaneous cerebrospinal-fluid reservoir. Preliminary report of 60 cases. N Engl J Med. 1968. 279: 1025-31
10. Rubenstein JL, Fridlyand J, Abrey L, Shen A, Karch J, Wang E. Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J Clin Oncol. 2007. 25: 1350-6
11. Sandberg DI, Bilsky MH, Souweidane MM, Bzdil J, Gutin PH. Ommaya reservoirs for the treatment of leptomeningeal metastases. Neurosurgery. 2000. 47: 49-54
12. Shapiro WR, Posner JB, Ushio Y, Chemik NL, Young DF. Treatment of meningeal neoplasms. Cancer Treat Rep. 1977. 61: 733-43
13. Witorsch P, Williams TW Jr, Ommaya AK, Utz JP. Intraventricular administration of amphotericin B. Use of subcutaneous reservoir in four patients with mycotic meningitis. JAMA. 1965. 194: 699-702
14. Yang S, Dai J, Zhang X, Jin Y. Intracerebral arachnoid cyst treated with Ommaya reservoir implantation in a patient younger than two years. J Craniofac Surg. 2014. 25: e378-80
Department of Neurosurgery, Shanghai Children's Medical Center Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai, People's Republic of China
Department Neurosurgery, UCLA Center for World Health at David Geffen School of Medicine, Tiverton Drive, Los Angeles, CA 90024, USA
Correspondence Address: Nan Bao Department Neurosurgery, UCLA Center for World Health at David Geffen School of Medicine, Tiverton Drive, Los Angeles, CA 90024, USA
How to cite this article: Bao N, Lazareff J. How I Do It: Management of spina bifida in a hospital in The People's Republic of China. Surg Neurol Int 23-Jul-2015;6:
We present our personal experience on patients with Spina Bifida. It is the result of having treated 1600 children for 12 years at Shanghai Children's Medical Center. We classify the cases on Spina Bifida Manifesta (myelomeningocele, myelocele, lypomyelomeningocele) or Spina Bifida Oculta (lipoma, dermal sinus and thickened filum terminale). For the former, we recommend surgery within 24â48 h after birth. For the latter we recommend preventive surgery months after birth. We acknowledge that the diameter of the spinal canal is a problem for large remnant lesions. In cases of myelomeningocele, we prefer to place the shunt and close the defect in the same procedure, it reduces the risks inherent to exposure to anesthesia, reduces hospital stay, and related costs. If there is a suspicious of infection, we do not place the shunt on the same procedure. The personal description of the preferred techniques for closure of the different defects is described.
There are several types of neural tube defects and each type can be divided into various subtypes. According to our experience in the treatment of nearly 1600 patients with different types of neural tube defects at the Neurosurgery Department of Shanghai Children's Medical Center over the past 12 years, we divided common neural tube defects into the following types:
Spina bifida manifesta
This type can be further divided into the following subtypes according to the pathological morphology:
Myelomeningocele
Patients with this subtype of neural tube defect have a mass on their back. The surface of the mass is a thin cyst wall and in some cases there is no skin [ Figure 1 ]. In other cases, the mass is covered by skin but the color is blue; there is no subcutaneous fat tissue, and the dermis shows scar-like degeneration and adheres directly to the cyst wall [ Figure 2 ]. The cyst wall is composed of the dura mater, arachnoid mater, pia mater, and deformed spinal cord, which protrudes outside the skin via the spinal defect. According to differences in the diseased segments and morphologies of spinal cord herniation, this subtype can be further divided into two subtypes. Type I is a herniation of the end of the spinal cord, in which the herniated spinal cord terminates at the roof of the bulging dura mater. It is commonly seen in the lumbosacral and sacral segments [Figures 1 and 2 ]. Type II is an omega-shaped (Ω-shaped) herniation of the spinal cord, in which the middle part of the herniated spinal cord makes up the roof of the bulging dura mater, but the end of the spinal cord is located in the distal spinal canal. This type is commonly seen in the lumbar and thoracolumbar segments [ Figure 3 ].
Figure 1
(a and b) Myelomeningocele. The tumor surface is a thin cyst wall, and the end of the spinal cord protrudes into the bulging dural sac via the spinal defect
Figure 2
(a) Myelomeningocele. The tumor surface is covered by skin, there is no subcutaneous fat tissue, the dermis shows a scar-like degeneration, and (b) the end of the spinal cord protrudes into the bulging dural sac via the spinal defect
Figure 3
(a and b) Myelomeningocele. There is a Ω-shaped protrusion of the spinal cord into the bulging dural sac
Neurological damage is very serious in patients with myelomeningocele. Most patients with type I myelomeningocele have bladder-sphincter dysfunction and most patients with type II myelomeningocele have lower extremity dysfunction, foot deformities, and even bladder-sphincter dysfunction and spinal deformities, etc., The incidence of Chiari malformation and hydrocephalus is as high as 99%, and these conditions are followed in incidence by syringomyelia, diastematomyelia, arachnoid cyst, etc.
Myelocele
Patients with this subtype of neural tube defect have a mass on their back. There is purple granulation at the center of the mass, which is surrounded by a thin cyst wall. The purple granulation is actually the Ω-shaped herniated spinal cord, which is directly exposed outside of the skin [ Figure 4 ].
Figure 4
(a) Myelocele. There is a purple granulation surface, and a Ω-shaped protrusion of the spinal cord is directly exposed outside the skin. (b) Sagittal MRI of the lesion
This subtype is commonly seen in the lumosacral, lumbar, and thoracolumbar segments is accompanied by the most serious neurological damage, and is associated with lower extremity dysfunction, foot deformities, bladder-sphincter dysfunction, spinal deformities, Chiari malformation, hydrocephalus, etc.
Lipomyelomeningocele
In this subtype the enlarged spinal cord protrudes dorsally via the defect in the spinal canal to form a mass that protrudes over the skin surface. The skin on the surface of the mass is intact. The mass contains subcutaneous fat tissue, cerebrospinal fluid (CSF), and spinal cord. The subcutaneous fat grows together with the herniated spinal cord and dura mater to form the cyst roof. Similar to myelomeningocele, this subtype can be divided into two subtypes according to the different morphologies of the herniated spinal cord. Type I is a herniation of the end of the spinal cord and is commonly seen in the lumbosacral and sacral segments [ Figure 5 ]. Type II is an Ω-shaped herniation of the spinal cord and is commonly seen in the lumbar and thoracolumbar segments [ Figure 6 ].
Figure 5
(a) Lipomyelomeningocele. The skin on the surface of a bulging dural sac is completely covered with subcutaneous fat tissue. (b) The end of the spinal cord protrudes into the bulging dural sac and grows together with the subcutaneous tissue to form the cyst roof
Figure 6
(a and b) Lipomyelomeningocele. There is a capillary hemangioma on the tumor surface and a skin depression in the sacrococcygeal region, an Ω-shaped protrusion of the spinal cord can be seen within the bulging dural sac, and subcutaneous fat grows into the spinal cord
Lipomyelomeningocele is a mass covered with normal skin, and there may be abnormal pigmentation or skin depression on the mass surface. Patients with lipomyelomeningocele may have varying degrees of lower extremity paralysis, foot deformities, gait abnormality, and bladder-sphincter dysfunction. Severe cases are often associated with Chiari malformation, hydrocephalus, cerebral dysplasia, hydromyelia, diastematomyelia, etc. Late symptoms include scoliosis, hydronephrosis, etc.
Simple meningocele
This subtype is characterized by bulging of the dura mater from the bone defect, and the cyst contains only CSF without spinal cord or cauda equina. If the cyst protrudes dorsally from the spinal canal, it is called a posterior simple meningocele [ Figure 7 ]. If the cyst protrudes ventrally from the spinal canal, it is called an anterior sacral meningocele [ Figure 8 ]. It was previously believed that simple meningocele usually has no neurological symptoms. However, more and more investigators have found that simple meningocele is associated with a very high rate of tethered spinal cord, which results in gradual neurological symptoms with increasing age. During the last 4 years of continuous observation, we found that in 37 of 38 cases (97%) simple meningocele was associated with other spinal cord diseases, such as tight filum terminale, arachnoid cyst, epidermoid cyst, tethered spinal cord due to fibrous bands, adhesion between the dura mater and the spinal cord or cauda equina, etc.
Figure 7
Posterior simple meningocele. There is no neural tissue in the bulging dural sac, the conus medullaris is located low at the level of L5, and there is associated fat degeneration
Figure 8
Anterior sacral meningocele. The cyst protrudes toward the ventral side of the sacrum and coccyx, the end of the spinal cord, which is located at the base of the bulging dural sac, sends out a band growing into the bulging dural sac, which results in spinal cord tethering
Occult spinal bifida
Skin in the affected area often has characteristic features including pigmentation, capillary hemangioma, skin depression, local hirsutism, small skin tags, etc. No significant symptoms can be observed in infants, and tethered spinal cord syndrome appears during the gradual development of the child after abnormal traction of the spinal cord occurs. It was reported that many patients have symptoms only after they grow up. Neural injury is mostly caused by the compression, traction, or increased tension of the spinal cord. If there is no surgical treatment, neural injury will become further aggravated and irreversible. Therefore, early diagnosis is very important for performing surgical intervention as soon as possible.
Occult spinal bifida is further divided into the following subtypes:
Spinal cord lipoma
A large amount of subcutaneous fat moves into the spinal canal via the spinal defect, and the dorsal dura mater is completely invaded by the subcutaneous lipoma and loses its normal structure. The lipoma enters the subdural space and grows with the lower located spinal cord. If the lipoma grows into the superficial part of the dorsal spinal cord, it is called a dorsal spinal cord lipoma [ Figure 9 ]. If the lipoma grows deep into the spinal cord unilaterally or bilaterally and even reaches the ventral side, it is called a ventral spinal cord lipoma [ Figure 10 ]. Hence, the spinal cord is compressed and pulled, which results in a tethered spinal cord.
Figure 9
Dorsal spinal cord lipoma. The lipoma grows into the superficial layer of the spinal cord
Figure 10
Ventral spinal cord lipoma. The lipoma grows deep into the spinal cord and even grows to the ventral side
A spinal cord lipoma is seen as a subcutaneous lipoma in the back with a capillary nevus or skin depression. Sometimes, it is only evident as a small skin tag. Affected patients may have varying degrees of bladder-sphincter dysfunction, lower extremity paralysis, foot deformities, and gait abnormality.
Spinal cord lipomas are commonly seen in the lumbosacral and sacrococcygeal segments, and are called lumbosacral spinal cord lipoma [ Figure 11 ] and sacrococcygeal spinal cord lipoma, respectively [ Figure 12 ]. These two types have slightly different pathological changes and the corresponding surgical procedures are also different. A spinal defect of a lumbosacral spinal cord lipoma is located in the lumbosacral segment, and the conus medullaris is located at the lumbosacral region or even lower. A subcutaneous lipoma invades the dura mater via the spinal defect and grows together with the spinal cord. Due to the lack of dorsal dura mater, bilateral residual dura tissues grow into the spinal cord inferior to the lipoma and lie superior to the bilateral nerve roots and the end of the conus medullaris. Therefore, the spinal cord is pulled downward and is tethered to the dura mater. Meanwhile, the lipoma grows cephalically and caudally within the spinal canal. In general, a cephalically growing lipoma enters the normal subdural space and grows into the dorsal part and/or one side of the spinal cord, while a caudally growing lipoma is mostly located outside the dura mater.
Figure 11
Lumbosacral spinal cord lipomas. (a) subcutaneous lipoma in the lumbosacral region. (b) MRI evidences that it grows into the spinal canas via the defects in the lumbosacral fascia, spinous process, dura mater and pia mater, and grows together with the lower located spinal cord
Figure 12
Sacrococcygeal spinal cord lipomas. (a) subcutaneous lipoma in the sacrococcygeal region. (b) MRI evidences that it grows into the dura mater via the sacral defect and grows together with the lower located spinal cord
Unlike a lumbosacral spinal cord lipoma, a sacrococcygeal spinal cord lipoma is located in the sacral canal. The subcutaneous lipoma in the sacrococcygeal region invades the dura mater via the sacral canal defect and grows together with the lower located spinal cord. Instead of terminating in the middle of the lumbosacral dural sac, the conus medullaris terminates in the distal end of the dural sac. Therefore, the cauda equina does not extend longitudinally from the end of the conus medullaris in accordance with normal anatomy, but extends obliquely downward from the ventral side of the conus medullaris. The lipoma is located in the superficial layer of the conus medullaris and grows into the conus medullaris. Differences in surgical procedures will be described in detail in the part on treatment.
Back dermal sinus
This subtype develops on the dorsal side of the cerebrospinal axis, at any site between the occiput and the sacrococcygeal region, most commonly in the lumbosacral region. The sinus may terminate outside or inside the dura mater. At the termination of the sinus is often a dermal cyst, which is located at the end of the spinal canal or grows into the spinal cord and causes tethered spinal cord [ Figure 13 ].
Figure 13
Back dermal sinus. (a) there is a pinprick-like hole in the skin. (b, c) MRI shows that a subcutaneous sinus enters the spinal canal via the dura mater, and the terminal is a dermal cyst, which grows form the outside into the spinal cord
It is seen as pinprick-like holes on the skin with peripheral abnormal hairs, pigmentation, or capillary hemangioma-like changes. Surgery should be performed as early as possible to prevent serious results such as secondary cyst infection, cerebrospinal meningitis, etc.
Diastematomyelia
This subtype can be further divided into two subtypes according to the presence or absence of clinical symptoms: Type I (presence of symptoms): There are two dural sacs and two spinal cords, that is, the spinal cord is divided into two parts. Each has its own dura mater and arachnoid mater and there is fibrous tissue, cartilage, or bone crest between the two parts, which causes tethered spinal cord [ Figure 14 ]. Type II (absence of symptoms): The spinal cord is divided into two parts at the diastemata, but these two parts share a common dura mater and arachnoid mater and there are no foreign bodies causing tethering, so most cases do not have clinical symptoms.
Figure 14
Diastematomyelia. (a) there is an abnormal hair bundle n the back. (b) Three dimensional CT shows that the bone crest grows into the spinal canal. (c) MRI show two spinal cords divided by a central bone crest
It is commonly seen in the thoracic and lumbar segments, and is seen as an abnormal hair clump in the center of the back. Ninety percent of patients with diastematomyelia have associated scoliosis.
Thickened filum terminale syndrome
When the terminal filament is invaded by fat and fibrous tissues, it degenerates and becomes thickened, consequently pulling down the spinal cord and causing neural symptoms. The neural symptoms caused by the tethered terminal filament are often relatively mild, and patients only have pain, enuresis, urinary urgency, fecal leakage, pes cavus, slight foot varus or valgus, etc. Location of the conus medullaris may be normal or lower [ Figure 15 ].
Figure 15
(a) MRI shows that the conus medullaris is located at the L2 level, and there is fat signal inside the distal end of the filum terminale. (b) intraoperative endoscopy shows fat degeneration in the filum terminale
Intradural lipoma
There is a localized fat accumulation in the subdural space without connection to the subcutaneous fat tissue in the back. The lipoma is often adhered to the dura mater on one side and located on the surface of the spinal cord on the other side. It also can grow into the spinal cord and cause tethered spinal cord [ Figure 16 ]. Smaller lipomas do not develop symptoms for life if they do not enlarge significantly as children grow.
Figure 16
A dorsal spinal cord lipoma compresses the spinal cord
Treatment
Aims: The goal of treatment is to improve neurological function and prevent further neural degeneration.
Ideal age and indication for surgery: Myelocele and myelomeningocele without skin covering may result in continuous CSF leakage due to the thin cyst wall. To reduce the risks of CSF leakage and infection, and subsequent continuous degeneration of neurological function, we often perform surgery to close the defect within 24–48 h after birth. For skin-covered neural tube defects, such as lipomyelomeningocele, elective surgery should be performed after considering the patient's age, body weight, general condition, and tolerance of the delicate spinal cord to outside intervention. Because the tethering and compression caused by the disease during the growth of the spinal cord may lead to further dysfunction, we perform surgery for patients with lipomyelomeningocele and spinal cord lipomas within 2–3 months after birth. However, the spinal canal is thinner in 2- to 3-month-old infants while the protruded spinal cord is abnormally thick, which makes complete reduction of the spinal cord difficult, produces extensive spinal cord adhesion, and increases the risk of subsequent tethered spinal cord. Therefore, the most suitable age for surgery in these children should be further investigated.
In patients with occult spinal bifida the disease is often detected a few years after birth based on manifestations including thickened filum terminal syndrome, diastematomyelia, and back dermal sinus. Surgery is often performed late in these patients.
Myelocele and myelomeningocele are associated with a high incidence of hydrocephalus. With regard to hydrocephalus requiring shunt surgery, there are differing opinions about whether shunt surgery should be performed together with surgeries for the original diseases or whether surgery should be carried out in two stages. We agree with those who advocate one-stage surgery because it allows the patients to undergo surgery and anesthesia only once, shortens the length of hospital stay, and reduces the medical cost. Because surgery for hydrocephalus is relatively less associated with problems, and CSF loss occurs during myelomeningocele repair, which may decrease the volume of the cerebral ventricle and make ventricular puncture become difficult, we perform shunt surgery after placing the patient in the supine position and then carry out myelomeningocele repair after turning the patient over to the prone position. To reduce the rate of complications including shunt occlusion and infection, we carry out strict decontamination of the surgical site, use very strict aseptic technique during surgery, minimize intraoperative loss of CSF, prevent cephalic spread of bloody CSF, reduce the time of wound exposure, etc., Of course, for patients with definite or potential infection of the central nervous system, myelomeningocele repair is done first and second-stage hydrocephalus shunt surgery is performed after the infection of the central nervous system is controlled.
Surgical principle: The spinal cord should be separated from the adhered lesion and the lesion removed to relieve spinal cord compression and tethering.
Surgical technique: Because the tumor surface is not covered by skin in patients with myelocele and myelomeningocele, the site of defect should be wrapped with saline-moistened gauze immediately after the patient is brought into the NICU to avoid drying and direct injury of the neural substrates due to exposure. The patient should be placed in a supine or lateral recumbent position and administered antibiotics intravenously.
Various measures should be used to prevent possible intraoperative hypothermia. For example, we increase the temperature of the operating room and put a heating pad underneath the child, avoid wrapping the abdomen and chest using wet towels during operation, and perform surgery as soon as possible according to the plan. The whole procedure should be carried out under microscope. The patient is placed in a prone position and a soft pad is placed underneath the lower abdomen to elevate the lower abdomen and buttock to the level of the head to maximally reduce CSF leakage during the procedure.
At the beginning of the surgery, an incision is made along the margin of exposed neural placode and the placode is separated from the peripheral tissue to get it back into the spinal canal. All the contents are trimmed including skin and granulation tissue on the placode. A neural tube resembling the spinal cord is then reconstructed, and the pia mater and arachnoid mater are drawn close to the midline from the bilateral margins of the placode and sutured. All the neural tissues are carefully protected and special attention is paid to electrocoagulation in order to avoid thermal burn injury to the placode, which may have residual function.
The dura mater is then dissected. One side of the dura mater is adjacent to the margin of the skin defect and is completely isolated from the skin. The dura mater is closed using 5-0 absorbable suture line without compressing the spinal cord.
Finally, the skin is sutured. If a patient has a large myelomeningocele, the large-area skin defect cannot be repaired by simple closure. Therefore, Z-shaped skin flaps are used or large-scale subcutaneous dissection is performed as far as both sides of the back to ensure a tension-free skin closure. Blunt finger dissection is carried out to avoid damaging large blood vessels. This kind of dissection can be performed quickly, which not only reduces bleeding but also ensures blood supply to the skin flap. Use of Z-shaped skin flaps or performing large-scale subcutaneous dissection guarantees direct skin suture for all patients.
In patients with a lipomyelomeningocele, after the cyst is separated, most of the time, half of the laminae of the vertebrae superior and inferior to the spina bifida is removed to sufficiently expose the base of the cyst. The normal dura mater is cut open to identify the relationship between the herniated spinal cord and myelomeningocele to avoid damaging the spinal cord when separating the cyst wall. An incision is made at the side of the apex of the cyst containing protruded spinal cord, the nerve within the cyst is carefully protected, and annular resection of the remaining parts is done under direct vision. Complete dissection is performed to release the spinal cord and neural fibers adhered to the cyst wall. Because a subcutaneous lipoma invades the spinal cord in cases with lipomyelomeningocele, the fat outside the spinal cord should be removed using a scissor and the fat inside the spinal cord should be removed using a micro scissor or an ultrasonic aspirator as much as possible to expose the layer of neural placode. Finally, interrupted suturing is performed for the split spinal cord, which is placed into the spinal canal. The redundant bulging dura mater is trimmed, and expanded suturing to the dural sac is done to prevent neural tissue compression and adhesion.
Surgical procedures for the two subtypes of myelomeningocele or lipomyelomeningocele are similar. For type I, the only requirement is to dissect and cut off the herniated distal end of the spinal cord from the bulging dura mater [ Figure 17 ]. For the Ω-shaped spinal cord herniation in type II, the herniated spinal cord cannot be cut off as is done in type I because it may break the spinal cord into two parts and result in irreversible neurological damage. The spinal cord should be separated from the bulging dural sac along the course of the spinal cord and placed back into the spinal canal [ Figure 18 ].
Figure 17
(a) MRI, (b) skin lesion, (c) cord tethered to the bottom of the canal and to the extradural fat, (d) cord untetehered
Figure 18
(a) MRI evidencing cord herniated in omega shape, (b) the bulging dura sac has been opened, (c) the spinal cord is separated from the sac, (d) the freed cord is about to be placed in the spinal canal
For simple meningocele, a fusiform incision should be made around the mass and dissection should be performed from the outside of the cyst wall to the neck of the cyst. The cyst wall should be cut open at the apex to explore the presence of neural tissue. According to the preoperative MRI findings, the bottom of the cyst cavity should be slightly expanded to explore the spinal canal, and find out whether there is fat degeneration of filum terminale, fibrous band tethering of the spinal cord, and adhesion between the dura mater and the spinal cord or the cauda equina. Corresponding tethering should be released. The redundant cyst wall should be trimmed and the dura mater sutured at the base. The par spinal muscle and fascia should be dissected around the laminar defect and the spinal defect covered using the reinforced suture technique.
Because there is no definite boundary between an intramural lipoma and normal spinal cord, complete resection of the lipoma is impossible. The aim of surgery is to dissect adhesion between the lipoma and the dura mater, reduce the volume of the lipoma, and release spinal cord tethering and compression to allow the reconstructed spinal cord to be suspended in the subarachnoid space satisfactorily.
The surgical technique for a spinal cord lipoma involves cutting open the dura mater from the cephalic normal site until the lipoma is completely exposed. The tumor membrane should be cut open and the lipoma should be gradually removed outside the spinal cord with a micro scissor. After most lipoma lesions outside the spinal cord are removed and the spinal cord is decompressed, the spinal cord should be slowly lifted from the ventral side of the spinal canal. At this point, the boundary of spinal cord is not yet isolated and the lipoma close to the spinal cord surface should not be removed in a hurry to avoid damaging the spinal cord below it. The spinal cord should be gently retracted to one side to expose the dura mater on the lateral side. The spinal cord should be dissected and cut off from both sides of the dura mater using a micro scissor in a cephalic to caudal direction to release the tethering and expose the spinal cord boundary. Then the fat and fibrous tissue should be further removed from the spinal cord surface safely, effectively, and maximally using the ultrasonic aspirator or CO2 laser knife until the layer of the neural plate is exposed. The small amount of fat tissue within the spinal cord should not be forcefully removed in order to protect spinal cord function.
Surgery for sacrococcygeal spinal cord lipoma is more difficult than surgery for lumbosacral spinal cord lipoma. In patients with lumbosacral spinal cord lipoma, the conus medullaris is located in the middle segment of the lumbosacral dura mater and the boundary is easily observed. As long as the normal dura mater is dissected at the caudal end of the lipoma and cut open cephalically, the conus medullaris can be identified and isolated from the dura mater. However, in patients with sacrococcygeal spinal cord lipoma, the conus medullaris is located in the distal end of the dural sac. The lipoma on the surface of the conus medullaris not only grows together with the conus medullaris, but also grows outside the sacral canal and connects with the normal fat tissue in the sacrococcygeal area without boundaries. Because the lipoma completely covers the conus medullaris and distal end of the dura mater, it is extremely difficult to identify the boundary between the conus medullaris and distal end of the dura mater during surgery and separate them. Our method is to diminish the fat from the cephalic end, lift the spinal cord from the ventral side after the fat becomes thinned, and then cut off the spinal cord from the dura mater. When the spinal cord boundary is exposed, the fat inside the spinal cord should be further removed in a cephalic to caudal direction, and the lipoma of the conus medullaris and tethering should then be treated. The key point is to accurately identify the boundary between the distal end of the dural sac and the conus medullaris in order to move toward the midline from both sides, dissect the conus medullaris, and release it from the end of the dural sac. If dissection toward the midline is performed too early, it will cut off and damage the conus medullaris, and if the dissection is carried out too late, it may cause disorientation and result in the manipulation being moved outside the caudal thecal sac or even the sacral canal, which not only cannot release the conus medullaris tethering, but also may damage the sacrococcygeal epidural spinal nerve. The fat on the surface of the conus medullaris should be diminished. When the fibrous fat layer is exposed, the dural sac end on the surface of the conus medullaris should be initially identified. The conus medullaris should be gently pulled aside at this point to further identify the distal end of the dural sac inside the sacral canal because the course of the dural sac end slants upward within the sacral canal. After the sacral sac end is reached, dissection should be carried out from both sides to the midline to completely isolate the conus medullaris from the end of the dural sac and entirely release the tethered spinal cord. After the boundary of the conus medullaris is totally exposed, the fibrous fat tissue on the surface of the conus medullaris should be removed safely and effectively and the neural plate reached.
Finally, the opened spinal cord should be repaired with interrupted suture to reduce the risk of postoperative adhesion between the dorsal side of the spinal cord and the suture site in the dura mater, and minimize the possibility of secondary tethering.
The key point of surgery for diastematomyelia is to remove the septum, regardless of bone, fat, or cartilage, because it is the cause of tethering. The bony septum outside the dura mater should be removed as much as possible using a ranger or small awl. In most cases, there are many blood vessels around the septum, which may cause massive bleeding if injured. The dura of the two spinal cords should be cut open. Often there are fibrous adhesions between the spinal cord and the dura mater at the site of the septum, and any such adhesions should be completely separated.
Surgery for thickened filum terminale syndrome should be performed via an incision between L4 and L5 or L5 and S1 spinouts processes. The dura mater and arachnoid mater should be cut open and the filum terminale identified according to midline location, yellow or silver change of filum’ color, disappearance of nodes of Rangier, and fat infiltration. The filum terminale should be separated from the peripheral nerve and slightly rotated to identify the presence of nerve adhesion on the ventral side. After electrocoagulation, 5 mm of filum terminale should be cut off as a specimen for pathological evaluation.
Surgery for a back dermal sinus requires complete removal of the dermal cyst and sinus inside and outside the spinal cord. It is necessary to identify the terminal of the sinus. Though sometimes a back dermal sinus that terminates on the surface of the dura mater is shown in imaging examinations, cutting open the dura mater is still needed for exploration because some tiny dermal cysts within the dura mater are often not shown on MRI.
Postoperative follow-up
Because some changes such as swelling caused by surgery disappears at least 1 month after surgery on MRI images, we often perform MRI 2–3 months after surgery to learn the postoperative status of the spinal cord. Close observation is carried out for patients with myelocele or myelomeningocele to identify the presence or absence of progressive aggravation of hydrocephalus. If the neurological function is stable, we regularly follow-up our patients 1, 3, and 5 years after surgery. We have a complete patient database and every patient is followed up by an experienced doctor. This may help doctors obtain first-hand information, learn the postoperative status of neurological function, find problems, and in time, adjust or improve surgical procedures.
How to cite this article: Lam S, Reddy GD, Lin Y, Jea A. Management of hydrocephalus in children with posterior fossa tumors. Surg Neurol Int 23-Jul-2015;6:
Case 1: A 2-year-old male with no prior medical history presented to the emergency room with a 3-week history of constant headache and daily vomiting. Computed tomography (CT) and subsequent magnetic resonance imaging (MRI) of the brain [Figure 1] showed a minimally enhancing mass in the fourth ventricle, which extended out through the foramen of Luschka on the left. There was associated supratentorial hydrocephalus. He had no evidence of spinal metastasis on MRI of the spine. There was no papilledema on the fundoscopic exam. He underwent placement of a right frontal external ventricular drain (EVD) and gross total resection of the tumor through a modified telovelar approach at the same time. The pathology was consistent with a grade II ependymoma. Postoperatively, the ventricular drain was unable to be weaned, and he underwent ventriculoperitoneal shunt placement without complication 1.5 weeks after initial surgery. He was eating and ambulatory after recovery. He went on to radiation therapy.
Figure 1
Magnetic resonance images of patient described in case 1. (a) Sagittal precontrast. (b) Axial fluid-attenuated inversion recovery. (c) Axial postcontrast
Case 2: A 9-year-old male with no prior medical history presented to an outside hospital emergency room with 2 weeks of progressive headaches and 1-day of vomiting. A CT of the head showed a posterior fossa mass. MRI of the brain [Figure 2] showed an enhancing fourth ventricular tumor with associated metastatic lesions throughout both cerebellar hemispheres and supratentorial hydrocephalus. There was no evidence of spinal metastasis. Fundoscopic exam was positive for papilledema. He underwent placement of a right frontal EVD and resection of the fourth ventricular mass through a modified telo-velar approach at the same time. The infiltrative lesions in the cerebellum were not resected. The pathology was consistent with medulloblastoma. Postoperatively, his EVD was weaned over the course of 2 weeks and removed. He did not require permanent cerebrospinal fluid diversion. He was discharged home after recovery and went on for adjuvant radiation therapy.
Figure 2
Magnetic resonance images of patient described in case 2. (a) Sagittal precontrast. (b) Axial fluid-attenuated inversion recovery. (c) Axial postcontrast
Central nervous system tumors are the most common solid tumors in children, and they predominantly occur in the posterior fossa.[7] Due to the anatomic relationships of these tumors to cerebrospinal fluid (CSF) drainage pathways, hydrocephalus is common, occurring in 71–90% of children with posterior fossa tumors.[11] Hydrocephalus after tumor resection occurs in 10–36% of cases,[24] with a worldwide average of 30%.[10]
The optimal management of hydrocephalus in a child with a posterior fossa tumor is a topic of debate.[13] The question of whether to place an external ventricular drain (EVD), insert a ventriculoperitoneal shunt (VPS), perform an endoscopic third ventriculostomy (ETV), or defer CSF diversion procedures before resective surgery depends on the clinical presentation and individual surgeon practice; there exists no class I evidence to guide management. In 2001, Sainte-Rose et al. reported that preoperative ETV was associated with a lower rate of postoperative hydrocephalus (27% vs. 6%) in a retrospective series of pediatric patients with posterior fossa tumors (n = 196).[11] Only a portion of these patients would have gone on to develop postresection hydrocephalus, so performing a preresection ETV in every case potentially exposes over 70% of patients to unnecessary surgery.[46]
Purported benefits of permanent preresection CSF diverting surgery, such as ETV of VPS other than the reduced incidence of postresection hydrocephalus, include the following: (1) Being able to delay resection surgery, thus avoiding resection under emergent conditions or allowing for preresection adjuvant therapy in certain circumstances;[3] (2) reducing the likelihood of needing external CSF diversion, which may carry risk of infection;[3] and (3) potentially reducing risk of postresection CSF leak or pseudomeningocele.[2] Purported disadvantages of permanent preresection CSF diversion surgery include the following: (1) Performance of a procedure that ultimately may not be clinically indicated, exposing patients to the risks of unnecessary surgery; (2) ETV may be less reliable in controlling intracranial pressure (ICP) and does not allow for ICP monitoring; and (3) no ability to externally drain spillage of blood products after the resection. The exact cause of postresection hydrocephalus is not completely characterized, with absorptive and obstructive processes implicated.
Ideally, we would be able to predict which patients will develop postresection hydrocephalus. The benefits of early CSF diversion could be captured while simultaneously avoiding the harm of subjecting patients to unnecessary procedures. Many groups have attempted to analyze retrospective data looking for clinical factors associated with a need for postoperative CSF diversion. Culley et al. (n = 117, 1976–1990) found that age <3 years, midline tumor location, subtotal resection, prolonged EVD requirement, cadaveric (vs. autologous) dural grafts, pseudomeningocele formation, and CSF infections were statistically significant factors associated with the need for postoperative shunt placement.[2] Due-Tønnessen and Helseth (n = 87, 1990–2003) found that patients with medulloblastoma and ependymoma had much higher rates of postoperative shunt requirement than astrocytomas.[4] Kumar et al. (n = 175, 1983–1993) found age <3, ependymoma/medulloblastoma tumor histology, and subtotal resection to be risk factors.[8] Santos de Oliveira et al. (n = 64, 1990–2006) found younger age, midline location, and greater ventricular index at presentation to be risk factors.[12] Morelli et al. (n = 160, 1989–2004) found medulloblastoma histology and severe preoperative hydrocephalus to be risk factors.[9] Bognár et al. (n = 180, 1990–2000) found younger age, tumor histology, and presence of EVD to be predictive of postoperative need for CSF diversion, but they found that tumor location, extent of resection, and postoperative CSF leak or pseudomeningocele were not predictive.[1]
In 2009, Riva-Cambrin et al. used a cohort of 343 patients to develop a clinical prediction rule for postresection hydrocephalus and validated it against another cohort of 111 patients from another institution in an attempt to identify high-risk patients who would benefit most from prophylactic ETV.[10] The group analyzed demographic, clinical, and radiographic factors. They performed stepwise multivariate regression to determine which factors were associated with a greater risk of needing CSF diversion after tumor resection and assigned point values reflecting the relative weights. The final scale is out of 10 points, with 3 points given for age <2 years, 1 point given for papilledema, 2 points given for moderate or severe hydrocephalus, 3 points given for cerebral metastases, and 1 point given for ependymoma, medulloblastoma, or dorsally exophytic brainstem glioma pathology predicted by preoperative radiology report. A score of >4 points was chosen as the cut-off for “high-risk.” Those with a score of 0–2 are predicted to have <20% chance of developing postresection hydrocephalus while the likelihood is >80% for those with a score of 7–10. High-risk (score 5–10) and low-risk (score 0–4) groups differed in posttest probabilities for developing postresection hydrocephalus by 48% (73% for high-risk, 25% for low-risk).[10] Foreman et al. later validated and modified Riva-Cambrin et al.’ predictive model, using fewer variables in a much smaller cohort (n = 99 patients): Age <2 years, moderate/severe hydrocephalus, preoperative tumor diagnosis per radiology report, and transependymal edema. These posterior fossa tumor patients were also stratified into high- and low-risk categories for development of postresection hydrocephalus.[5]
There exists no class I evidence in the literature to guide the management of hydrocephalus in children with posterior fossa tumors. It is possible to draw guidance from the extant data highlighted above. As the overall incidence of postresection hydrocephalus is typically 30%, any anticipated benefit should be weighed against exposing the patient to more surgery or permanent shunt implantation. It is noted that in lower resource settings, there may be other considerations, including the cost of care, access to the operating room and need to minimize the number of surgeries. In our practice, in cases where there is no hydrocephalus on presentation, preresection CSF diversion is not done. In cases where there is symptomatic hydrocephalus on presentation, preresection EVD, VPS or ETV should be applied as clinically appropriate. EVD is favored for its advantages of expedient placement, external control over drainage perioperatively, and egress of resection-related blood and protein products. In cases where the child possesses multiple described risk factors for the development of postresection hydrocephalus, preresection prophylactic CSF diversion may be considered. Overall, close observation is recommended, with a preference for expectant management, rather than prophylactic surgery, and postresection definitive CSF diversion procedures undertaken only as clinically necessary.
1. Bognár L, Borgulya G, Benke P, Madarassy G. Analysis of CSF shunting procedure requirement in children with posterior fossa tumors. Childs Nerv Syst. 2003. 19: 332-6
2. Culley DJ, Berger MS, Shaw D, Geyer R. An analysis of factors determining the need for ventriculoperitoneal shunts after posterior fossa tumor surgery in children. Neurosurgery. 1994. 34: 402-7
3. Di Rocco F, Jucá CE, Zerah M, Sainte-Rose C. Endoscopic third ventriculostomy and posterior fossa tumors. World Neurosurg. 2013. 79: S18.e15-9
4. Due-Tønnessen BJ, Helseth E. Management of hydrocephalus in children with posterior fossa tumors: Role of tumor surgery. Pediatr Neurosurg. 2007. 43: 92-6
5. Foreman P, McClugage S, Naftel R, Griessenauer CJ, Ditty BJ, Agee BS. Validation and modification of a predictive model of postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr. 2013. 12: 220-6
6. Fritsch MJ, Doerner L, Kienke S, Mehdorn HM. Hydrocephalus in children with posterior fossa tumors: Role of endoscopic third ventriculostomy. J Neurosurg. 2005. 103: 40-2
7. Johnson KJ, Cullen J, Barnholtz-Sloan JS, Ostrom QT, Langer CE, Turner MC. Childhood brain tumor epidemiology: A brain tumor epidemiology consortium review. Cancer Epidemiol Biomarkers Prev. 2014. 23: 2716-36
8. Kumar V, Phipps K, Harkness W, Hayward RD. Ventriculo-peritoneal shunt requirement in children with posterior fossa tumours: An 11-year audit. Br J Neurosurg. 1996. 10: 467-70
9. Morelli D, Pirotte B, Lubansu A, Detemmerman D, Aeby A, Fricx C. Persistent hydrocephalus after early surgical management of posterior fossa tumors in children: Is routine preoperative endoscopic third ventriculostomy justified?. J Neurosurg. 2005. 103: 247-52
10. Riva-Cambrin J, Detsky AS, Lamberti-Pasculli M, Sargent MA, Armstrong D, Moineddin R. Predicting postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr. 2009. 3: 378-85
11. Sainte-Rose C, Cinalli G, Roux FE, Maixner R, Chumas PD, Mansour M. Management of hydrocephalus in pediatric patients with posterior fossa tumors: The role of endoscopic third ventriculostomy. J Neurosurg. 2001. 95: 791-7
12. Santos de Oliveira R, Barros Jucá CE, Valera ET, Machado HR. Hydrocephalus in posterior fossa tumors in children. Are there factors that determine a need for permanent cerebrospinal fluid diversion?. Childs Nerv Syst. 2008. 24: 1397-403
13. Schijman E, Peter JC, Rekate HL, Sgouros S, Wong TT. Management of hydrocephalus in posterior fossa tumors: How, what, when?. Childs Nerv Syst. 2004. 20: 192-4
How to cite this article: Niknejad HR, Samii A, Shen S, Samii M. Huge familial colloid cyst of the third ventricle: An extraordinary presentation. Surg Neurol Int 23-Jul-2015;6:
Background:Since the use of computed tomography and magnetic resonance imaging, colloid cysts (CCs) are discovered more frequently and subsequently their true incidence exceeds the numbers previously estimated. In 1986, the first familial case was reported in two identical twin brothers. To date, a total of 17 of these cases have been reported, all differing in the pattern of affected family members.
Case Description:Here, we describe a unique presentation of a familial case and review the relevant literature on CCs and their natural history to improve our understanding of these cases.
Conclusion:Familial CC can present in various patterns, sizes, and forms. A genetic factor is likely to be responsible in these cases, and further research is warranted to clarify this phenomenon.
Colloid cysts (CC's) of the third ventricle are benign intra-cranial cysts that account for approximately 1% of all intra-cranial tumors. Considering the intraventricular lesions only, they comprise up to 20% of all tumors, and they are the most common mass found in the third ventricle.[410222729] The true incidence of CC's can only be estimated, because of the large cohort of asymptomatic individuals. Usually, the patients are middle-aged though the cysts can occur at virtually all ages.[917] The first case report dates from 1858 by Wallmann, who described the lesion both clinically and pathologically.[48] From there, it took several decades and progress in the field of neurosurgery to allow Dandy to successfully remove a CC in 1921.[13] Since this achievement, these lesions were considered curable as surgery offered a definite solution for the disease. Nowadays, CC's are not considered to be a neoplasm, rather a developmental malformation composed of a fibrous outer layer, internally bordered by a ciliated or mucus-producing epithelium. It is the activity of this very epithelium that determines their growth and expansion, leading to the capacity to cause the neurological decline. Although they are known for their slow growth and indolent character, their strategic position at the foramen of Monro not seldom gives them malicious traits.[36] This is often inconsistent with their usually small size. More precisely, they most often lay in the anterior part of the third ventricle, between the forniceal columns, obliterating the foramen of Monro and causing hydrocephalus.
They mainly present with signs and symptoms related to hydrocephalus such as headache and nausea, which are too unspecific to pinpoint an exact etiology. On the other hand, we have the silent cases diagnosed incidentally by means of magnetic resonance imaging (MRI) or computed tomography (CT).[34] On imaging, these structures are seen as spherical isodense to hyperdense lesions on CT. On T2-weighted MRI, they usually appear hyperintense, and unless the surrounding fibrous capsule is vascular, they do not enhance on contrast agents. Finally CC's may incidentally be found at autopsy, as in the case of Dr. Harvey Cushing.[15]
To date, a total of 17 familial cases has been reported.[1235672425303233353738434447] We would like to replenish the series with a case that is, unique, as it concerns two middle-aged nontwin brothers with large to gigantic CC's. The cyst sizes of all familial case reports are listed in Table 1. In our case descriptions, we also report the technical notes of how we surgically managed the cases.
Table 1
Overview of the literature on familial CC of the third ventricle
In 2006, a 43-year-old Saudi man was referred to our institution, the International Neuroscience Institute in Hannover, for the treatment of a cystic intra-cranial lesion. During the week before admission, the primary complaint was sudden onset headaches accompanied by episodes of vomiting. At the time of evaluation, the headaches were described as constant, very severe and diffuse. Sensorimotor evaluation showed mild numbness in the right hand, with a slight weakness of the hand extensors on the right side. A CT-scan performed in the country of origin revealed a cystic lesion that appeared to be a CC of the third ventricle. On T1-weighted gadolinium-enhanced images a 25 mm × 25 mm × 20 mm measuring cystic lesion was seen [ Figure 1 ] causing mild obstructive hydrocephalus. The mass itself was also strongly contrast enhanced, leading us to believe the cyst wall contained some aberrant vessels.
The patient underwent a right fronto-dorsal parasagittal craniotomy for a microsurgical extirpation of the lesion through a transcallosal approach. Total removal of the cyst and its colloidal content, together with its aberrant vascular steel was achieved.
Figure 1
First case. (a) Preoperative gadolinium enhanced T1-weighted axial magnetic resonance imaging, showing an in homogenously enhancing mass in the third ventricle causing mild hydrocephalus. (b) Postoperative axial computed tomography-scan showing complete removal of the lesion and absence of hydrocephalus
Histological examination of the biopsy specimens confirmed the diagnosis of a CC. Surprisingly, the vascular steel consisted of a cluster of markedly abnormal arteries together with tortuous and dilated veins, in accordance with our pathological diagnosis of an arteriovenous malformation (AVM).
Postoperatively the patient recovered quickly and there were no complications. At discharge, the headaches and vomiting were no longer present and the patient was free of any other neurological deficits.
Second case
After a 6-year interval, the 39-year-old brother of our first patient was admitted to our clinic with a 1-month history of vertigo together with episodes of nausea and vomiting. One week prior to admission the patient had lost consciousness after having an acute onset headache and was transported to a local hospital. His past medical history and familial history were unremarkable for brain diseases. However, a detailed familial history was not obtained, nor were any other family members examined at our center. A CT-scan was performed showing hydrocephalus and a huge cystic lesion in the third ventricle. In the acute setting, he was treated by means of a ventricular drain. Preoperative gadolinium-enhanced T1-weighted MRI showed a huge, 52 mm × 42 mm × 39 mm measuring, hypointensive cystic lesion in the third ventricle that had progressively expanded upward to the corpus callosum, displacing the septum pellucidum and bulging in the lateral ventricles [ Figure 2 ]. Diffusion tensor imaging (DTI) fiber tracking visualized a pronounced displacement of the fornices to the right [ Figure 3 ]. Therefore, we planned a left frontal parasagittal craniotomy to gain access to the lesion through a transcallosal approach from the opposite side of the forniceal structures. The surgery was performed under intraoperative MRI control with DTI fiber tracking in order to visualize the forniceal structures. After puncturing the cyst and aspirating the entire glue like the content, the capsule was partially removed. To restore the liquor flow a large and wide-opening was made in the cyst wall ventro-cranially, to ensure outflow to the lateral ventricles. Moreover, a second opening was created caudally in the area of the lamina terminalis, to form a connection between the third ventricle and the suprasellar cisterns. The foraminae of Monro were identified, as they were displaced more ventrally, and their patency was secured. Intra-operative MRI showed total extirpation of the cyst content as well as the artificial “cysto-cisternal” and “cysto-ventricular” openings. Finally, endoscopic inspection was performed to ensure hemostasis. There were no postoperative complications and no memory deficits were detected on subsequent serial examinations. Histology confirmed the diagnosis of a CC, showing a single layered AE1/AE3 positive ciliated epithelium.
Figure 2
Second case. (a) Preoperative gadolinium enhanced T1-weighted axial magnetic resonance imaging, showing a gigantic cystic lesion in the third ventricle causing hydrocephalus. Note the enhancing vessels on the cyst wall. Artifact is due to the occipital venticuloperitoneal-shunt. (b) Postoperative axial computed tomography-scan showing extirpation of the cyst content and no signs of increased intra-cranial pressure
Figure 3
Second case diffusion tensor imaging (DTI). Preoperative DTI fiber tracking. Coronal (a) and axial (b) images showing a pronounced displacement of the fornices inferiorly and to the right. The artifact is due to the ventriculoperitoneal-shunt
Ever since their discovery, CC's have remained a curious clinicopathological entity. More than a century ago Sjovall presumed the cysts originated out of the paraphysis cerebri, an evanescent vestigial embryonic structure.[ 42 ] This theory held state until the advent of the term “neuroepithelial cyst” in 1929 by Fulton and Bailey. They discussed the presence of cilia and certain cyst contents in their specimens along with the variability in the location of the cysts, holding both a pathological and an anatomical argument against a paraphyseal origin. In 1955 Kappers partially restored Sjovall's theory by explaining that an ectopic location of the cyst results from inclusion of peripheral paraphyseal “anlagen,” which is of ependymal origin and may arise along variable locations along the ventricular axis.[ 26 ] Electron microscopy allowed Coxe and Luse to subscribe an ependymal epithelial origin in 1964 though their findings were based on a single case.[ 11 ] One year later Shuangshoti et al. introduced their theory of neuroepithelial origin.[ 41 ] Based on a review of the literature combined with embryological and comparative anatomical studies, they classified the paraphysis as the extraventricular choroid plexus. The suggestion that both choroid plexus and ependyma are derived from a common neuroepithelium can balance the arguments Fulton and Bailey held against the paraphyseal origin, somewhat unifying the former theories. Besides this, some authors have proposed an extraneural origin, namely out of the ectopic endodermal tissue. The argument for this theory is the similarity of the cyst epithelium to the respiratory mucosa, described by Tsuchida et al.[ 45 ] Concurrently Ho and Garcia found the presence of ciliated cells and goblet cells upon ultrastructural analysis of their specimens. In their arrangement, the cells were interconnected by desmosomes and met the criteria of an endodermal lineage.[ 23 ] These findings have led to the supposition that CC's and Rathke's cleft cysts may share the same pathophysiology and represent comparable lesions at different locations.[ 18 ] Still the true origin of the CC's remains a matter of debate.
Colloid cysts and genetics
Another approach of trying to clarify the mechanisms involved in the development of familial CC's is the search for a genetic abnormality that could lead to an inheritable disease. Interestingly in this respect, insights into the function of “paired”-like homeodomain transcription factor (Prop1) in the development of the Rathke's pouch, the pituitary primordium, have been described in mice. Prop1 seems to have a crucial role in cell proliferation and differentiation and thus in the organogenesis and function of the pituitary gland. A dysregulation in Prop1 expression is correlated with an increased susceptibility for pituitary tumors and Rathke's cleft cysts. The latter were found in 40% of the alpha glycoprotein subunit (αGSU)-Prop1 transgenic mice, which express a high level of Prop1 under the αGSU promoter (gain of function).[ 12 ] In humans the Prop1 gene fulfills the same function, and several loss-of-function mutations have been known to cause dysfunction and cystic dysplasia of the pituitary.[ 49 51 ] By analogy with Rathke's cleft cysts it is likely that a genetic factor is involved in causing CC's as well. Especially since these clinical conditions may constitute the same entity. The idea of an inheritable genetic factor involved in the pathophysiology of familial CC's of the third ventricle has already risen. This assumption is made, because of the improbable statistical chance of co-occurrence in first-degree relatives (1:1011).[ 46 ] Despite the fact that this phenomenon can occur in a very variable way with regard to the family members affected, an autosomal dominant inheritance seems to be the most likely form of inheritance.[ 17 32 44 ] Our case shows that huge cysts, earlier described solely in individual cases,[ 20 50 ] can also arise in kinship. In addition, we found an AVM in the first case. Previous reports have mentioned the solitary occurrence of AVM in the third ventricle.[ 8 21 39 ] On the other hand, some intra-cranial anomalies have been described in association with CCs, such as craniopharyngioma,[ 28 ] xanthogranuloma,[ 19 31 ] astrocytoma,[ 16 ] and agenesis of the corpus callosum.[ 14 ] To the best of our knowledge, it is the first time that an AVM is reported in association with a CC of the third ventricle. These ascertainments, revealing an additional variable factor in how this phenomenon may present, advocate in favor of a developmental malformation.[ 40 ] Yet it remains worthwhile to invest in research to pinpoint a genetic defect that would offer an explanation for the cases observed.
1. Aggarwal A, Corbett A, Graham J. Familial colloid cyst of the third ventricle. J Clin Neurosci. 1999. 6: 520-2
2. Ahmed SK, Stanworth PA. Colloid cysts of the third ventricle in identical twins. Br J Neurosurg. 2002. 16: 303-7
3. Akins PT, Roberts R, Coxe WS, Kaufman BA. Familial colloid cyst of the third ventricle: Case report and review of associated conditions. Neurosurgery. 1996. 38: 392-5
4. Antunes JL, Louis KM, Ganti SR. Colloid cysts of the third ventricle. Neurosurgery. 1980. 7: 450-5
5. Bavil MS, Vahedi P. Familial colloid cyst of the third ventricle in non-twin sisters: Case report, review of the literature, controversies, and screening strategies. Clin Neurol Neurosurg. 2007. 109: 597-601
6. Bengtson BP, Hedeman LS, Bauserman SC. Symptomatic neuroepithelial (colloid) cysts of the third ventricle. A unique case report in nontwin brothers. Cancer. 1990. 66: 779-85
8. Britt RH, Silverberg GD, Enzmann DR, Hanbery JW. Third ventricular choroid plexus arteriovenous malformation simulating a colloid cyst. Case report. J Neurosurg. 1980. 52: 246-50
9. Buchsbaum HW, Colton RP. Anterior third ventricular cysts in infancy. Case report. J Neurosurg. 1967. 26: 264-6
10. Camacho A, Abernathey CD, Kelly PJ, Laws ER Jr. Colloid cysts: Experience with the management of 84 cases since the introduction of computed tomography. Neurosurgery. 1989. 24: 693-700
11. Coxe WS, Luse SA. Colloid cyst of third ventricle. an electron microscopic study. J Neuropathol Exp Neurol. 1964. 23: 431-45
13. Dandy WE. Diagnosis, localization and removal of tumours of the third ventricle. Bull Johns Hopkins Hosp. 1922. 33: 188-9
14. del Carpio-O’Donovan R, Cardinal E. Agenesis of the corpus callosum and colloid cyst of the third ventricle: Magnetic resonance imaging of an unusual association. Can Assoc Radiol J. 1990. 41: 375-9
15. Fulton JF. Harvey Cushing: A Biography. Spring-field: Charles C Thomas. 1946. p. 713-4
16. Gelabert M, Bollar A, Martinez R, Allut AG. Coincidence of a frontal lobe astrocytoma and colloid cyst of the third ventricle. Neurochirurgia (Stuttg). 1991. 34: 69-70
17. Gemperlein J. Paraphyseal cysts of the third ventricle. Report of two cases in infants. J Neuropathol Exp Neurol. 1960. 19: 133-4
18. Graziani N, Dufour H, Figarella-Branger D, Donnet A, Bouillot P, Grisoli F. Do the suprasellar neurenteric cyst, the Rathke cleft cyst and the colloid cyst constitute a same entity?. Acta Neurochir (Wien). 1995. 133: 174-80
19. Hadfield MG, Ghatak NR, Wanger GP. Xanthogranulomatous colloid cyst of the third ventricle. Acta Neuropathol. 1985. 66: 343-6
20. Hamlat A, Casallo-Quiliano C, Saikali S, Adn M, Brassier G. Huge colloid cyst: Case report and review of unusual forms. Acta Neurochir (Wien). 2004. 146: 397-401
21. Heafner MD, Duncan CC, Kier EL, Ment LR, Scott DT, Kolaski R. Intraventricular hemorrhage in a term neonate secondary to a third ventricular AVM. Case report. J Neurosurg. 1985. 63: 640-3
22. Hernesniemi J, Romani R, Dashti R, Albayrak BS, Savolainen S, Ramsey C. Microsurgical treatment of third ventricular colloid cysts by interhemispheric far lateral transcallosal approach – Experience of 134 patients. Surg Neurol. 2008. 69: 447-53
23. Ho KL, Garcia JH. Colloid cysts of the third ventricle: Ultrastructural features are compatible with endodermal derivation. Acta Neuropathol. 1992. 83: 605-12
24. Ibrahim AW, Farag H, Naguib M, Ibrahim E. Neuroepithelial (colloid) cyst of the third ventricle in identical twins. Case report. J Neurosurg. 1986. 65: 401-3
25. Joshi SM, Gnanalingham KK, Mohaghegh P, Wilson A, Elsmore A. A case of familial third ventricular colloid cyst. Emerg Med J. 2005. 22: 909-10
26. Kappers JA. The development of the paraphysis cerebri in man with comments on its relationship to the intercolumnar tubercle and its significance for the origin of cystic tumors in the third ventricle. J Comp Neurol. 1955. 102: 425-509
27. Kelly R. Colloid cysts of the third ventricle; analysis of twenty-nine cases. Brain. 1951. 74: 23-65
28. Klein MR. Craniopharyngioma and tumor of the III ventricle: Exeresis of both tumors. Rev Neurol. 1994. 76: 21-
29. Little JR, MacCarty CS. Colloid cysts of the third ventricle. J Neurosurg. 1974. 40: 230-5
30. Mathiesen T, Grane P, Lindgren L, Lindquist C. Third ventricle colloid cysts: A consecutive 12-year series. J Neurosurg. 1997. 86: 5-12
31. Matsushima T, Fukui M, Kitamura K, Soejima T, Ohta M, Okano H. Mixed colloid cyst-xanthogranuloma of the third ventricle. A light and electron microscopic study. Surg Neurol. 1985. 24: 457-62
32. Nader-Sepahi A, Hamlyn PJ. Familial colloid cysts of the third ventricle: Case report. Neurosurgery. 2000. 46: 751-3
33. Partington MW, Bookalil AJ. Familial colloid cysts of the third ventricle. Clin Genet. 2004. 66: 473-5
34. Pollock BE, Huston J. Natural history of asymptomatic colloid cysts of the third ventricle. J Neurosurg. 1999. 91: 364-9
35. Romani R, Niemelä M, Korja M, Hernesniemi JA. Dizygotic twins with a colloid cyst of the third ventricle: Case report. Neurosurgery. 2008. 63: E1003-
36. Ryder JW, Kleinschmidt-DeMasters BK, Keller TS. Sudden deterioration and death in patients with benign tumors of the third ventricle area. J Neurosurg. 1986. 64: 216-23
37. Sadeghi S, Sharifi G, Aliasgari A. Familial colloid cyst of the third ventricle: A case report and review of the literature. Med J Islam Repub Iran. 2003. 17: 267-9
38. Salaud C, Hamel O, Buffenoir-Billet K, Nguyen JP. Familial colloid cyst of the third ventricle: Case report and review of the literature. Neurochirurgie. 2013. 59: 81-4
39. Shahhal I, Dayes LA. A case of arteriovenous malformation of the third ventricle: A clinical presentation and special features. Bull Clin Neurosci. 1984. 49: 13-22
41. Shuangshoti S, Roberts MP, Netsky MG. Neuroepithelial (colloid) cysts: Pathogenesis and relation to choroid plexus and ependyma. Arch Pathol. 1965. 80: 214-24
42. Sjovall E. Uber eine Ependymcyste embryonalen characters (paraphyse.) im dritten Hirnventrikel met todlichem Ausgang?. Beitr Pathol Anat. 1910. 47: 248-69
43. Socin HV, Born J, Wallemacq C, Betea D, Legros JJ, Beckers A. Familial colloid cyst of the third ventricle: Neuroendocrinological follow-up and review of the literature. Clin Neurol Neurosurg. 2002. 104: 367-70
44. Stoodley MA, Nguyen TP, Robbins P. Familial fatal and near-fatal third ventricle colloid cysts. Aust N Z J Surg. 1999. 69: 733-6
45. Tsuchida T, Hruban RH, Carson BS, Phillips PC. Colloid cysts of the third ventricle: Immunohistochemical evidence for nonneuroepithelial differentiation. Hum Pathol. 1992. 23: 811-6
46. Vandertop WP, Gosselaar PH, Nesselrooij B. Three sisters with colloid cyst of third ventricle. Lancet. 1995. 346: 643-4
47. Vandertop WP. Familial colloid cyst of the third ventricle: Case report and review of associated conditions. Neurosurgery. 1996. 39: 421-
48. Wallman H. Eine Colloidcyste im dritten Hirnventrikel und ein Lipom in Plexes Choroides. Virchous Arch. 1858. 11: 385-8
49. Wu W, Cogan JD, Pfäffle RW, Dasen JS, Frisch H, O’Connell SM. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. 1998. 18: 147-9
50. Yüceer N, Baskaya M, Gökalp HZ. Huge colloid cyst of the third ventricle associated with calcification in the cyst wall. Neurosurg Rev. 1996. 19: 131-3
51. Zygmunt-Górska A, Starzyk J, Adamek D, Radwanska E, Sucharski P, Herman-Sucharska I. Pituitary enlargement in patients with PROP1 gene inactivating mutation represents cystic hyperplasia of the intermediate pituitary lobe. Histopathology and over 10 years follow-up of two patients. J Pediatr Endocrinol Metab. 2009. 22: 653-60
How to cite this article: Kalimullah J, Humza Sohail AM A, Shahjehan RD, Siddique S, Bari ME. Neurofibromatosis type 2 patient presenting with medulloblastoma. Surg Neurol Int 07-Oct-2015;6:
Background:Neurofibromatosis type 2 (NF2) is an autosomal dominant syndrome with a frequency of 1 in 25,000 live births and a penetrance of almost 100% by the sixth decade of life. The main tumors occurring in NF2 patients are bilateral vestibular schwannomas, other peripheral, cranial and spinal nerve schwannomas, intracranial and intraspinal meningiomas, ependymomas, and gliomas.
Case Description:We report the case of a 6-year-old boy who presented with a 1-month history of nausea and recurrent vomiting. Physical examination was positive for ataxic gait and left-sided facial nerve palsy. Family history was positive for NF2 in the patient's father and paternal uncle. Magnetic resonance imaging brain revealed a solid enhancing lesion arising from the right cerebellar cortex, which was effacing the fourth ventricles and causing hydrocephalus. Craniotomy and excision of the lesion were performed. Histopathology report confirmed the diagnosis to be desmoplastic medulloblastoma. Based on the patient's subsequent history and family history, he was diagnosed to be a case of NF2.
Conclusion:This is the first case of medulloblastoma occurring in a patient with NF2 and raises the possibility of an association between medulloblastoma and NF2.
Keywords: Association, brain neoplasm, medulloblastoma, neurofibromatosis type 2
Neurofibromatosis type 2 (NF2) is an autosomal dominant syndrome with a frequency of 1 in 25,000 live births and a penetrance of almost 100% by the sixth decade of life.[2] It has a variable presentation, resulting from the mutation in the tumor suppressor gene merlin, which is located on chromosome 22q, and is a predominantly intracranial condition with its characteristic bilateral vestibular schwannomas.[15] The other main tumors occurring in NF2 patients are other peripheral, cranial and spinal nerve schwannomas, intracranial and intraspinal meningiomas, ependymomas and gliomas.[6] Four large clinical studies have been conducted which confirm the aforementioned clinical picture.[4101112] These tumors are mostly benign and grow slowly, but their location within the central nervous system can cause great morbidity and mortality.[9]
Clinical presentation of most patients with NF2 includes hearing loss which is usually unilateral, with or without tinnitus.[6] NF2 is diagnosed clinically after a patient fulfills a predefined criteria.[5]
Herein, we report a case NF2 presenting with recurrent vomiting and headache that was diagnosed with medulloblastoma. This, to the best of our knowledge, is the first case of medulloblastoma occurring in an NF2 patient.
A 6-year-old boy of average height and weight presented to the ER in 2003 with complaints of repeated vomiting and headache for 1-month. He was a student of a local public school, his past medical history being unremarkable; his immunization status was complete, and there were no known allergies. In family history, the patient's father and uncle (father's brother) were known to have NF2. On physical examination, he was vitally stable. Positive findings included left-sided facial nerve palsy and ataxic gait.
After a series of initial investigations, magnetic resonance imaging (MRI) brain was done. The report described a solid mass arising from the right cerebellar cortex, which was isointense to the gray matter on T1-weighted images and hypo to isointense to the gray matter on T2-weighted images. Postcontrast images showed an intense enhancement, which was almost homogenous. The mass was causing effacement of the fourth ventricles with dilatation of the third and lateral ventricles. The vertical height of the lesion was 4.2 cm; AP dimension was 4 cm, and the transverse diameter was 4.8 cm. There was no evidence of intracranial hemorrhage. Gray and white matter signals of supratentorial brain were within normal limits. No midline structural defect was seen. The differential diagnoses of medulloblastoma or astrocytoma were made.
The patient underwent craniotomy and excision of the lesion with the insertion of a ventriculoperitoneal shunt. The histopathology report described a malignant infiltrating tumor present in sheets [Figure 1]. The tumor was composed of small oval to indented basophilic cells, exhibiting nuclear hyperchromasia [Figures 1 and 2]. Extensive necrosis [Figure 3] and karyorrhexis were identified with areas of hemorrhage. Brisk mitotic figures were also seen [Figure 4]. The tumor cells showed positivity for immunohistochemical stain CD 56 [Figure 5]. Focal positivity for immunohistochemical stain glial fibrillary acidic protein was also seen [Figure 6]. Based on these findings a final diagnosis of desmoplastic medulloblastoma was made.
Figure 1
Patternless sheets of primitive appearing neoplastic cells with hyperchromatic nuclei with neuropil
Figure 2
Primitive appearing neoplastic cells with hyperchromatic nuclei, scant cytoplasm, and indistinct cell borders
Figure 3
Tumor with areas of necrosis as indicated by the arrow (H and E, ×20)
Figure 4
Neoplastic cells showing prominent mitotic figures as pointed out by the arrow (H and E, ×40)
Figure 5
Tumor cells showing positivity for immunohistochemical stain CD56
Figure 6
Tumor cells showing focal positivity for immunohistochemical stain glial fibrillary acidic protein
The patient underwent metastatic workup which was negative. After discharge, he was kept under close follow-up. Pediatric oncology team was taken on board and after discussing the case at the tumor board meeting, a multidisciplinary approach was taken and both chemotherapy and radiotherapy were administered.
In 2003, he was found to have a 1.0 cm × 1.5 cm skin colored nodule on this left forearm, which was excised with histopathology of the lesion revealing benign peripheral nerve sheath tumor.
In 2004, during a routine MRI scan the patient was found to have a cerebellar lesion for which he underwent craniotomy and excision of the lesion. Biopsy of the excised specimen revealed postchemotherapy and radiotherapy gliosis. He was then again kept under close follow-up. He complained of vision disturbances in 2006 and after an ophthalmological exam, was diagnosed to have right eye cataract for which he underwent phacoemulsification and insertion of the intraocular lens.
Till 2010, his routine follow-up MRI scans showed no significant pathological changes. However, in 2010, he again started complaining of vision disturbances and ophthalmological examination revealed right-sided posterior capsular opacification and left-sided cataract. He underwent left capsulectomy.
Over the span of next 3 years the patient remained well however, in 2013, he was again brought to Aga Khan University Hospital with complaints of headache, vomiting, and gait disturbances. MRI brain showed right superior frontal, inferior frontal, parasagittal and left posterior parietal meningiomas; excision of the lesions was performed.
Considering his case history and his family history, a diagnosis of NF2 was considered for him. A neurologist was taken on board; the NIH criterion was applied, and the patient was diagnosed to have NF2.
After the surgery in 2013, the patient has not been reported any symptoms. The follow-up MRI scans have not revealed any positive findings.
NF is a genetic disorder of the nervous system. It became widely recognized in the 19th century,[1] but it has a wide pictorial history that traces back to the 13th century.[3] The neuromas of NF were first described, in 1849.[3] Von Recklinghausen's is credited with the discovery of NF and coined the name of this disorder, in 1882.[3] Research on NF increased between 1909 and 1990 after Joseph Merick, the famous Elephant Man, was erroneously diagnosed with NF1.[3]
NF is considered to have two distinct types, NF1 and NF2.[14] NF1 has more peripheral manifestations and NF2 carries predominantly central manifestations. Over the past two decades, our knowledge of the genetics and management of NF has dramatically increased. The two forms of NF have been shown to be two distinct entities, both at clinical and molecular levels.[14] NF also has other rarer forms with outlying phenotypes and atypical presentations. These unconventional and atypical forms have been delineated and include hereditary spinal NF, schwannomatosis, familial intestinal NF, autosomal dominant “café au lait” spots alone, Watson syndrome, autosomal dominant “neurofibromas” alone, Noonan syndrome and the syndrome of multiple naevi, multiple schannomas, and multiple vaginal leiomyomas.[14]
NF2 is often a devastating autosomal dominant disorder which until recently was confused with its more common namesake NF1.[6] NF2 is a multiple neoplasia syndrome that results from a mutation in NF2 tumor suppressor gene on chromosome 22.[2] Affected patients carry a dominant loss of function mutation of the merlin gene on chromosome 22. Merlin is a cytoskeletal protein that functions as a tumor suppressor by facilitating E-cadherin-mediated contact inhibition.
NF2 inevitably develops schwannomas, typically affecting both the vestibular nerves, resulting in hearing loss and deafness. Most of the patients present with hearing loss which is unilateral at onset and may be accompanied or preceded by tinnitus.[7] Vestibular schannomas may also cause dizziness or imbalance as first symptoms. Nausea, vomiting or true vertigo are rare symptoms, occurring more commonly in the later stages.[7] The other tumors seen in NF2 are schwannomas of other cranial, spinal, and peripheral nerves. Furthermore, NF2 is associated with intracranial and intraspinal meningiomas and low-grade central nervous system malignancies like ependymomas.[7] As mentioned previously, both meningiomas and schwannomas were seen in our patient. Ophthalmic manifestations are also present in NF2 and include reduced juvenile cataracts, as seen in our patient, and retinal hamartomas.[8] About 70% of NF2 cases have skin tumors with intracutaneous plaque like lesions or more deep-seated tumors.[7]
Medulloblastoma is an aggressive posterior fossa brain tumor. Although medulloblastoma has been reported in patients with NF1, one study found the prevalence of posterior fossa tumors in NF1 to be 0.83%, there is no reported case of medulloblastoma occurring in a patient with NF2.[13]
To the best of our knowledge, this is the first case of medulloblastoma in a patient with NF2. The literature review conducted for this report could not find any case report or research study relating medulloblastoma and NF2. This raises the possibility of an association between these two disease entities and shows that even NF2 can also have this posterior fossa brain stem tumor.
1. Ahn MS, Jackler RK, Lustig LR. The early history of the neurofibromatosis. Evolution of the concept of neurofibromatosis type 2. Arch Otolaryngol Head Neck Surg. 1996. 122: 1240-9
2. Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z. Neurofibromatosis type 2. Lancet. 2009. 373: 1974-86
3. Brosius S. A history of von Recklinghausen's NF1. J Hist Neurosci. 2010. 19: 333-48
4. Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V. A clinical study of type 2 neurofibromatosis. Q J Med. 1992. 84: 603-18
5. Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V. A genetic study of type 2 neurofibromatosis in the United Kingdom. II. Guidelines for genetic counselling. J Med Genet. 1992. 29: 847-52
6. Evans DG, Sainio M, Baser ME. Neurofibromatosis type 2. J Med Genet. 2000. 37: 897-904
7. Evans DG. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J Rare Dis. 2009. 4: 16-
8. Feucht M, Griffiths B, Niemüller I, Haase W, Richard G, Mautner VF. Neurofibromatosis 2 leads to higher incidence of strabismological and neuro-ophthalmological disorders. Acta Ophthalmol. 2008. 86: 882-6
9. Han F. Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am J Hum Genet. 1996. 59: 331-42
10. Kanter WR, Eldridge R, Fabricant R, Allen JC, Koerber T. Central neurofibromatosis with bilateral acoustic neuroma: Genetic, clinical and biochemical distinctions from peripheral neurofibromatosis. Neurology. 1980. 30: 851-9
11. Mautner VF, Lindenau M, Baser ME, Hazim W, Tatagiba M, Haase W. The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery. 1996. 38: 880-5
12. Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N. Neurofibromatosis 2 (NF2): Clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet. 1994. 52: 450-61
13. Pascual-Castroviejo I, Pascual-Pascual SI, Viaño J, Carceller F, Gutierrez-Molina M, Morales C. Posterior fossa tumors in children with neurofibromatosis type 1 (NF1). Childs Nerv Syst. 2010. 26: 1599-603
14. Ruggieri M. The different forms of neurofibromatosis. Childs Nerv Syst. 1999. 15: 295-308
15. Uppal S, Coatesworth AP. Neurofibromatosis type 2. Int J Clin Pract. 2003. 57: 698-703
How to cite this article: Lam S, Reddy GD, Mayer R, Lin Y, Jea A. Eosinophilic granuloma/Langerhans cell histiocytosis: Pediatric neurosurgery update. Surg Neurol Int 07-Oct-2015;6:
A 23-month-old female was admitted to the neurosurgery service with a 3-month history of a progressively enlarging neck mass. There was associated redness, swelling, and tenderness to palpation, but no neurological deficits on examination. A noncontrast computed tomography (CT) scan of the neck and magnetic resonance imaging (MRI) with contrast showed an osteolytic contrast-enhancing lesion primarily involving the C2 posterior elements, with a compressive circumferential epidural component extending from C2 to C5 [ Figure 1 ]. A skeletal survey was negative for any other osseous lesions. She underwent C2 to C5 laminectomy with partial resection of the lesion without complication. Pathology was consistent with Langerhans cell (LC) histiocytosis (LCH). She was discharged home several days after her operation and subsequently started outpatient chemotherapy with cytarabine.
Figure 1
Images for the patient in case 1. (a) Computed tomography of the cervical spine without contrast. Sagittal (top) and axial (bottom) images. (b) Magnetic resonance imaging of the upper spine with contrast. Sagittal (left) and axial (right) images
Case 2
A 17-year-old male was admitted to the neurosurgery service with a 6-week history of a progressively enlarging scalp mass. There were tenderness and intermittent bleeding from the ulcerated lesion, with no focal neurologic deficits on examination. A noncontrast CT scan of the head and MRI brain, with and without contrast, showed a large transosseous contrast-enhancing lesion of the right frontal bone [ Figure 2 ]. CT venography showed compression of the adjacent superior sagittal sinus. Surgery with gross total resection of the lesion was performed. Pathology was consistent with LCH. The patient was discharged home the day after his operation. He subsequently started outpatient chemotherapy with cytarabine.
Figure 2
Images for the patient in case 2. (a) Computed tomography head without contrast. Coronal brain window (top), coronal bone window (middle), and three-dimensional skull reconstruction (bottom) images. (b) Magnetic resonance imaging brain. Coronal postcontrast T1-weighted image (top) and sagittal T2-weighted (bottom) images
Overview and nosology of Langerhans cell histiocytosis
LCH is a rare heterogeneous illness characterized by the proliferation of dendritic cells with LCH morphology. LCH refers to a spectrum of diseases, from a localized lesion to a diffuse multiorgan pathology.[ 10 27 43 ] It can affect any organ system, but most commonly involves the skeletal system (80%). Other commonly affected sites include the skin (33%), pituitary gland (25%), liver, spleen, lungs, and brain.[ 23 ] The most common named subtype, eosinophilic granuloma (EG), refers to a benign localized LCH, most commonly of the bone.[ 43 ] Other named subtypes include Hand–Schüller–Christian disease, a multifocal LCH classically characterized by exophthalmos, diabetes insipidus, and osteolytic skull lesions, and Letterer–Siwe disease, a diffuse systemic LCH clinically manifested by skin rash, hepatosplenomegaly, and pancytopenia with an acute fulminant course. A congenital self-limited form, named Hashimoto–Pritzker disease, has also been described.[ 24 29 ] In 1953, Lichtenstein grouped all of these diseases under the name “histiocytosis X,” a term that has since been replaced by LCH.[ 24 34 ]
Etiology and pathophysiology
As mentioned above, the proliferation of a myeloid-derived precursor dendritic cell with the characteristics of a LC is what characterizes LCH.[ 11 19 ] One theory on the pathophysiology of LCH is based on recently published data that LCH cells express CD40 at high levels, leading to the overactivation of CD40L + T-lymphocytes. The subsequent release of cytokines leads to further recruitment of LC progenitor cells, as well as to the local destruction of bone, fibrosis, and necrosis.[ 1 13 18 ]
The central debate on the pathogenesis of LCH is whether it is a reactive immune response or a neoplasm. Support for the neoplastic theory includes the monoclonality of the pathologic cells (though this also can be seen with reactive immune processes),[ 1 49 ] the up-regulation of pro-cell proliferation and anti-apoptotic proteins (c-Myc, H-ras, Bcl-2) in LCH samples,[ 47 ] the up-regulation of Ki-67 (a marker of proliferation),[ 5 47 ] the finding of activated BRAF mutations in a majority of samples, and the clinical response of BRAF (V600E) disease to vemurafenib.[ 4 22 ] Support for the theory that LCH represents a dysfunctional immune response includes data showing the up-regulation of genes that lead to T-cell activation and recruitment in LCH lesions,[ 2 ] an increase of T-regulatory cells in the blood of LCH patients in comparison with controls,[ 48 ] and a lack of evidence of gross genomic or karyotypic aberration in LCH biopsy samples.[ 9 ] Of note, no antigenic trigger has been identified, and evidence points against a viral etiology.[ 26 36 ]
Epidemiology
EG that affects the bone is the most common subtype of LCH, representing an estimated 60–80% of cases. EG can be single or multifocal; it most commonly affects the calvarium, but can also present in the vertebrae, ribs, long bones, and mandible.[ 12 29 37 ] EG primarily affects children under the age of 15, with an estimated incidence of <1 per 100,000.[ 10 27 ]
Clinical presentation, workup, and diagnosis
Radiography demonstrates a sharply demarcated osteolytic lesion of the underlying bone.[ 37 43 ] EG of the skull presents as a gradually enlarging scalp mass.[ 43 ] EG of the vertebral body in children most commonly presents with pain. A neurologic deficit is uncommon even in cases of progression to vertebra plana.[ 6 20 ] EG of the orbital bone may present as proptosis with an accompanying mass mimicking malignancy.[ 15 ] EG may also occur intracranially, typically in the hypothalamic-pituitary axis with associated endocrinopathies.[ 25 ] Radiographically, LCH lesions avidly enhance with contrast on both CT and MR and demonstrate increased uptake in fluorine-18–labeled fluorodeoxyglucose (FDG)-positron emission tomography (PET).[ 33 ] They appear hypointense on T1-weighted MRI sequences and iso- to hyperintense on T2.[ 10 21 ]
The differential for an osteolytic mass is broad and includes neoplastic processes such as metastatic lesions, primary bone tumors, including osteosarcoma or Ewing's sarcoma, neuroblastoma, rhabdomyosarcoma, lymphoma and primitive neuroectodermal tumors. It also includes infections such as osteomyelitis or abscesses, fibrous dysplasia, and cystic lesions such as aneurysmal bone cysts or dermoid cysts. Vascular processes, such as venous lakes, hemangiomas, and angiomatosis, are also in the differential, as well as developmental anomalies such as neurenteric cysts and encephaloceles. Tissue biopsy is necessary for definitive diagnosis. The classic radiographic presentation of vertebra plana (not shown in our more dramatic Case 1) is not considered pathognomonic of LCH, as complete vertebral collapse can be seen with other diagnoses such as Ewing's sarcoma and infection.
Histopathologically, LCH lesions show a proliferation of LC-type cells in a milieu of lymphocytes, macrophages, and eosinophils. In 1987, the Histiocyte Society established the diagnostic criteria for a diagnosis of LCH, which required the identification of CD1a on immunohistochemistry or Birbeck granules on electron microscopy.[ 24 ] Immunohistochemistry for Langerin, a protein enriched in and necessary for the formation of Birbeck granules, is an acceptable modern alternative to electron microscopy.[ 1 23 ] Recent work suggests Birbeck granules are part of the endosomal recycling system and may be involved with loading CD1a (a protein similar to major histocompatibility complex-I) with glycoprotein antigens for presentation to T-cells.[ 35 ]
Workup should include a systemic survey to identify any other potential sites of involvement, as management recommendations and prognosis vary depending on the number and type of organs involved in LCH.[ 17 ] The Euro Histio network published guidelines for the workup of LCH in 2013 based on the available review of the literature and recommends the following laboratory tests: Complete blood count with differential, complete metabolic panel (electrolytes, [blood urea nitrogen] BUN/creatinine and liver function tests), erythrocyte sedimentation rate, coagulation studies (prothrombin time/international normalization ratio, partial thromboplastin time, fibrinogen), chest X-ray, and radiographic skeletal survey.[ 23 ] The Histiocyte Society recommends the same laboratory workup, with the addition of urine specific gravity and abdominal ultrasound.[ 38 ] Endocrine labs (follicle-stimulating hormone/luteinizing hormone, thyroid stimulating hormone, growth hormone, cortisol) are indicated if there is any suspicion of pituitary involvement. MRI is useful for the detection of bone marrow or soft-tissue involvement, and a fast whole-body T2-STIR protocol is a possible screening test.[ 30 44 ] PET has been shown to be superior to bone scans for lesion detection, but it exposes children to higher radiation doses.[ 44 ] Mueller et al. found MRI had high sensitivity (81%), but FDG-PET had a higher specificity (76%) for the detection of LCH lesions.[ 40 ]
Treatment
LCH of the spine is rare. Treatment patterns in the literature include observation, complete surgical excision with fixation, and radiotherapy. Bertram et al. reviewed the literature of spine EG (n = 53) and found that most cases resolved without treatment. Immobilization and observation are recommended in cases without spinal instability or neurological deficit.[ 6 ] Similar findings have been reported by Raab et al. (n = 14) and Yeom et al. (n = 23).[ 46 50 ] LCH spares the endochondral plates; thus, recovery of vertebral heights is possible if the endochondral tissues are not disturbed with surgery or radiation.[ 28 46 ] Surgical excision, followed by segmental fusion and internal fixation, is indicated in cases of spinal instability, a neurological deficit from compression of the spinal cord or spinal nerves, or marked noncompliance with immobilization/external bracing.[ 28 ] Radiotherapy is no longer recommended for spine LCH, except in emergent cases of spinal cord compression.[ 1 23 ]
Solitary EG of the calvarial vault without invasion into neurological structures has a favorable prognosis. These lesions typically are managed with surgical curettage or excision.[ 11 23 28 ] Small case series reported by Oliveira et al. (n = 4) and by De Angulo et al. (n = 8) found that these lesions fully resolved without intervention after 6–19 months;[ 11 42 ] however, a major limitation of these reports is the lack of diagnostic certainty in the absence of tissue analysis. There is no Class I evidence for the management of these lesions, and treatment decisions typically are made on a case-by-case basis. On one hand, overly aggressive surgical excision may be associated with prolonged healing and deformity, particularly with large lesions. For this reason, the European Histiocyte Society recommends avoidance of surgical excision in lesions >5 cm in children.[ 23 ] On the other hand, observation alone also carries risk, since the differential diagnosis of LCH includes aggressive cancers and infection and the disease may progress to pathologic fracture or encroachment on neurological structures.[ 3 ] One option is to perform a biopsy or partial resection for definitive diagnosis, followed by observation or a conservative treatment option, such as an intralesional injection of steroids or interferon, or systemic treatments with indomethacin or bisphosphonates. These treatments have been shown to be efficacious in the literature;[ 14 16 41 ] however, the results must be interpreted in the context of a disease that potentially is self-resolving.
The Histiocyte Society recommends more aggressive treatment in cases of multifocal bone disease or disease that involves “CNS-risk” sites (odontoid, vertebrae with intraspinal soft-tissue extension, facial bones, skull base, orbit, oral cavity).[ 1 38 ] In these types of LCH, the risk of recurrence is high (30–50%), as is the risk of invasion into neurological tissue or development of neurologic sequelae (40%) such as endocrinopathies, diabetes insipidus, and parenchymal brain disease.[ 38 ] For LCH of this type, the Histiocyte Society recommends systemic treatment with prednisone and vinblastine for 12 months.[ 38 ] Recent studies, however, have shown less toxicity and lower recurrence rates in patients treated with cytarabine.[ 8 ] There is no Class I evidence or guidelines regarding surgical treatment for these lesions. Historically, lesions that invade the dura or compress neurological tissues and are located in accessible sites have undergone surgical excision.[ 7 29 31 ] In cases where surgical excision would be impossible or excessively morbid, systemic chemotherapy and/or injections of steroids or interferon may be a preferential alternative.[ 3 ] In the past, radiation therapy was used as an adjunctive treatment; however, it is no longer recommended due to concerns over the possible long-term effects of radiotherapy on the developing brain and spine.[ 1 23 ]
Prognosis
In a large study from South Korea (n = 603), 5-year overall survival was 99.8% in those with single-system LCH, 98.4% for multisystem LCH without risk organ involvement, and 77% for multisystem LCH with risk organ involvement.[ 32 ] Similar findings have been found by other groups in Japan (n = 91) and Italy (n = 121).[ 39 45 ] In a large study from the Mayo Clinic (n = 263), recurrence rates in those with single-bone LCH was 7%, and the appearance of new bone lesions was 14%.[ 31 ] As mentioned above, the Histiocyte Society reports a high rate of recurrence (30–50%) among those with “CNS-risk” lesions.[ 38 ]
LCH describes a heterogeneous mixture of pathologies and should be on the differential for any osteolytic soft-tissue mass. Definitive diagnosis requires tissue biopsy, and treatment options vary from observation to resection and chemotherapy. Prognosis is good, and recurrence rates are low, particularly for patients with single bone lesions.
1. Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: Current concepts and treatments. Cancer Treat Rev. 2010. 36: 354-9
2. Allen CE, Li L, Peters TL, Leung HC, Yu A, Man TK. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared to epidermal Langerhans cells. J Immunol. 2010. 184: 4557-67
3. Azouz EM, Saigal G, Rodriguez MM, Podda A. Langerhans’ cell histiocytosis: Pathology, imaging and treatment of skeletal involvement. Pediatr Radiol. 2005. 35: 103-15
5. Bank MI, Rengtved P, Carstensen H, Petersen BL. Langerhans cell histiocytosis: An evaluation of histopathological parameters, demonstration of proliferation by Ki-67 and mitotic bodies. APMIS. 2003. 111: 300-8
6. Bertram C, Madert J, Eggers C. Eosinophilic granuloma of the cervical spine. Spine (Phila Pa 1976). 2002. 27: 1408-13
7. Binning MJ, Brockmeyer DL. Novel multidisciplinary approach for treatment of Langerhans cell histiocytosis of the skull base. Skull Base. 2008. 18: 53-8
8. Cantu MA, Lupo PJ, Bilgi M, Hicks MJ, Allen CE, McClain KL. Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS One. 2012. 7: e43257-
9. da Costa CE, Szuhai K, van Eijk R, Hoogeboom M, Sciot R, Mertens F. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009. 48: 239-49
10. D’Ambrosio N, Soohoo S, Warshall C, Johnson A, Karimi S. Craniofacial and intracranial manifestations of Langerhans cell histiocytosis: Report of findings in 100 patients. AJR Am J Roentgenol. 2008. 191: 589-97
11. De Angulo G, Nair S, Lee V, Khatib Z, Ragheb J, Sandberg DI. Nonoperative management of solitary eosinophilic granulomas of the calvaria. J Neurosurg Pediatr. 2013. 12: 1-5
12. Denaro L, Longo UG, Papalia R, Di Martino A, Maffulli N, Denaro V. Eosinophilic granuloma of the pediatric cervical spine. Spine (Phila Pa 1976). 2008. 33: E936-41
13. Egeler RM, Favara BE, van Meurs M, Laman JD, Claassen E. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: Abundant expression of cytokines relevant to disease and treatment. Blood. 1999. 94: 4195-201
15. Erly WK, Carmody RF, Dryden RM. Orbital histiocytosis X. AJNR Am J Neuroradiol. 1995. 16: 1258-61
16. Farran RP, Zaretski E, Egeler RM. Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol. 2001. 23: 54-6
17. Gadner H, Minkov M, Grois N, Pötschger U, Thiem E, Aricò M. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013. 121: 5006-14
18. Geissmann F, Lepelletier Y, Fraitag S, Valladeau J, Bodemer C, Debré M. Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood. 2001. 97: 1241-8
19. Geissmann F. Histiocytosis and the Mononuclear Phagocyte System. Proceedings of the 24th Annual Meeting of the Histiocyte Society, October 1-3, Berlin, Germany. 2008. p.
20. Greenlee JD, Fenoy AJ, Donovan KA, Menezes AH. Eosinophilic granuloma in the pediatric spine. Pediatr Neurosurg. 2007. 43: 285-92
21. Gunny R, Clifton A, Al-Memar A. Spontaneous regression of supratentorial intracerebral Langerhans’ cell histiocytosis. Br J Radiol. 2004. 77: 685-7
22. Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013. 121: 1495-500
23. Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R. Langerhans cell histiocytosis (LCH): Guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013. 60: 175-84
24. . Histiocytosis syndromes in children. Writing Group of the Histiocyte Society. Lancet. 1987. 1: 208-9
26. Jeziorski E, Senechal B, Molina TJ, Devez F, Leruez-Ville M, Morand P. Herpes-virus infection in patients with Langerhans cell histiocytosis: A case-controlled sero-epidemiological study, and in situ analysis. PLoS One. 2008. 3: e3262-
27. Jubran RF, Marachelian A, Dorey F, Malogolowkin M. Predictors of outcome in children with Langerhans cell histiocytosis. Pediatr Blood Cancer. 2005. 45: 37-42
28. Karagoz Guzey F, Bas NS, Emel E, Alatas I, Kebudi R. Polyostotic monosystemic calvarial and spinal Langerhans’ cell histiocytosis treated by surgery and chemotherapy. Pediatr Neurosurg. 2003. 38: 206-11
29. Kasper EM, Aguirre-Padilla DH, Alter RY, Anderson M. Histiocytosis X: Characteristics, behavior, and treatments as illustrated in a case series. Surg Neurol Int. 2011. 2: 57-
30. Kellenberger CJ, Epelman M, Miller SF, Babyn PS. Fast STIR whole-body MR imaging in children. Radiographics. 2004. 24: 1317-30
31. Kilpatrick SE, Wenger DE, Gilchrist GS, Shives TC, Wollan PC, Unni KK. Langerhans’ cell histiocytosis (histiocytosis X) of bone. A clinicopathologic analysis of 263 pediatric and adult cases. Cancer. 1995. 76: 2471-84
32. Kim BE, Koh KN, Suh JK, Im HJ, Song JS, Lee JW. Clinical features and treatment outcomes of Langerhans cell histiocytosis: A nationwide survey from Korea histiocytosis working party. J Pediatr Hematol Oncol. 2014. 36: 125-33
33. Lee HJ, Ahn BC, Lee SW, Lee J. The usefulness of F-18 fluorodeoxyglucose positron emission tomography/computed tomography in patients with Langerhans cell histiocytosis. Ann Nucl Med. 2012. 26: 730-7
34. Lichtenstein L. Histiocytosis X; integration of eosinophilic granuloma of bone, Letterer-Siwe disease, and Schüller-Christian disease as related manifestations of a single nosologic entity. AMA Arch Pathol. 1953. 56: 84-102
35. Mc Dermott R, Ziylan U, Spehner D, Bausinger H, Lipsker D, Mommaas M. Birbeck granules are subdomains of endosomal recycling compartment in human epidermal Langerhans cells, which form where Langerin accumulates. Mol Biol Cell. 2002. 13: 317-35
36. McClain K, Jin H, Gresik V, Favara B. Langerhans cell histiocytosis: Lack of a viral etiology. Am J Hematol. 1994. 47: 16-20
37. Meyer JS, De Camargo B. The role of radiology in the diagnosis and follow-up of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998. 12: 307-26
39. Morimoto A, Ikushima S, Kinugawa N, Ishii E, Kohdera U, Sako M. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: Results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer. 2006. 107: 613-9
40. Mueller WP, Melzer HI, Schmid I, Coppenrath E, Bartenstein P, Pfluger T. The diagnostic value of 18F-FDG PET and MRI in paediatric histiocytosis. Eur J Nucl Med Mol Imaging. 2013. 40: 356-63
41. Munn SE, Olliver L, Broadbent V, Pritchard J. Use of indomethacin in Langerhans cell histiocytosis. Med Pediatr Oncol. 1999. 32: 247-9
42. Oliveira M, Steinbok P, Wu J, Heran N, Cochrane D. Spontaneous resolution of calvarial eosinophilic granuloma in children. Pediatr Neurosurg. 2003. 38: 247-52
43. Park SH, Park J, Hwang JH, Hwang SK, Hamm IS, Park YM. Eosinophilic granuloma of the skull: A retrospective analysis. Pediatr Neurosurg. 2007. 43: 97-101
44. Phillips M, Allen C, Gerson P, McClain K. Comparison of FDG-PET scans to conventional radiography and bone scans in management of Langerhans cell histiocytosis. Pediatr Blood Cancer. 2009. 52: 97-101
45. Postini AM, Andreacchio A, Boffano M, Pagano M, Brach Del Prever A, Fagioli F. Langerhans cell histiocytosis of bone in children: A long-term retrospective study. J Pediatr Orthop B. 2012. 21: 457-62
46. Raab P, Hohmann F, Kühl J, Krauspe R. Vertebral remodeling in eosinophilic granuloma of the spine. A long-term follow-up. Spine (Phila Pa 1976). 1998. 23: 1351-4
47. Schouten B, Egeler RM, Leenen PJ, Taminiau AH, van den Broek LJ, Hogendoorn PC. Expression of cell cycle-related gene products in Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2002. 24: 727-32
48. Senechal B, Elain G, Jeziorski E, Grondin V, Patey-Mariaud de Serre N, Jaubert F. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med. 2007. 4: e253-
49. Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH. Langerhans’-cell histiocytosis (histiocytosis X) – A clonal proliferative disease. N Engl J Med. 1994. 331: 154-60
50. Yeom JS, Lee CK, Shin HY, Lee CS, Han CS, Chang H. Langerhans’ cell histiocytosis of the spine. Analysis of twenty-three cases. Spine (Phila Pa 1976). 1999. 24: 1740-9
How to cite this article: Singh I, Rohilla S, Kumar P, Sharma S. Spinal dorsal dermal sinus tract: An experience of 21 cases. Surg Neurol Int 07-Oct-2015;6:
Background:Spinal dorsal dermal sinus is a rare entity, which usually comes to clinical attention by cutaneous abnormalities, neurologic deficit, and/or infection. The present study was undertaken to know the clinical profile of these patients, to study associated anomalies and to assess the results of surgical intervention.
Methods:Medical records of 21 patients treated for spinal dorsal dermal sinus from September 2007 to December 2013 were reviewed.
Results:We had 21 patients with male: female ratio of 13:8. Only 2 patients were below 1-year of age, and most cases (15) were between 2 and 15 years (mean age = 8.2 years). Lumbar region (11 cases) was most frequently involved, followed by thoracic (4 cases), lumbosacral, and cervical region in 3 patients each. All of our patients presented with neurological deficits. Three patients were admitted with acute meningitis with acute onset paraplegia and had intraspinal abscess. The motor, sensory, and autonomic deficits were seen in 14, 6, and 8 patients, respectively. Scoliosis and congenital talipes equinovarus were the common associated anomalies. All patients underwent surgical exploration and repair of dysraphic state and excision of the sinus. Overall, 20 patients improved or neurological status stabilized and only 1 patient deteriorated. Postoperative wound infection was seen in 2 cases.
Conclusions:All patients with spinal dorsal dermal sinuses should be offered aggressive surgical treatment in the form of total excision of sinus tract and correction of spinal malformation, as soon as diagnosed.
Spinal dorsal dermal sinus tract (DST) is a rare congenital dysraphism that occurs in approximately one in every 2500 live births.[17121718] It includes a tract lined by epithelium, which traverse for a variable depth into the underlying structures and in many instances, terminate within the thecal sac.[23] They are seen more frequently at the extremes of neuraxis with the majority of spinal DSTs occurring in the lumbosacral region.[67917] Spinal DSTs may have diverse and occasionally serious presentations; in fact, many cases come to clinical attention by neurologic deficit and/or infectious complications including life-threatening conditions such as meningitis.[6] In addition, DSTs are frequently associated with other anomalies of the central nervous system such as tethered cord, inclusion tumors, and split cord malformations (SCMs).[26] So despite its benign external appearance, it may harbor great risks to the patients’ health if not timely addressed. The neurological examination is reported to be normal in the early childhood. However, as the age increases, there is more chance of neurological deficit, which tends to be more profound. There are few published series in literature which emphasize mainly the mode of presentation, radiological findings, associated anomalies and treatment; however, the symptom wise outcome is not studied in detail.[136121718] The present study was undertaken to know the clinical profile, associated anomalies and detailed symptom wise outcome of the patients presenting with spinal DST.
This is a retrospective study conducted in Pt. B.D. Sharma University of Health Sciences, Rohtak from September 2007 to December 2013. Medical records of all patients treated for spinal DST were reviewed. Information regarding patients’ demographic variables, type of presentation, symptoms, physical examination, radiological and surgical findings, and histopathological evaluation were collected. Magnetic resonance imaging (MRI) was the investigation of choice and was performed in all cases. MRI revealed the relationship of the dermal sinus to the dural sac and also gave information regarding associated abnormalities in the cord like dysraphic state of spine or inclusion tumor.
Surgical intervention
The aim of surgery was to excise the sinus tract completely and to correct the dysraphic state in the same sitting. Surgery was performed in all cases through midline incision with encircling the sinus. DST was followed through the subcutaneous tissue and muscle layer sinus tract was traced until its end and excised completely. The course of DST was invariably rostral through the incompletely formed lamina or underneath the normal lamina. After doing the laminectomy, dura was opened in all cases irrespective of end of DST. In cases where DST was intradural, part of dura encircling the DST was excised. Intraspinal pathologies like SCM were dealt accordingly that is, dermoid and epidermoid were decompressed or excised; myelocele and lipomeningomyelocele were repaired; drainage of abscess in intramedullary abscess, removal of arachnoid adhesion in arachnoiditis and detethering of the cord was done in case of tethered cord. Those patients presenting with infectious complications were managed with appropriate antibiotics and then after recovery surgery for resection of DST, and correction of associated anomalies was performed. Postoperative follow-up ranged from 6 months to 5 years (mean - 2.8 years).
Records of total 21 patients were analyzed, of which 13 were male, and 8 were female. Patients’ age on admission ranged from 9 months to 15 years (mean - 8.2 years). Every patient underwent a detailed neurological examination and a complete radiological workup to delineate any underlying/associated spinal abnormalities. DST was located most frequently in lumbar region (11 cases) [Figure 1], followed by thoracic (4 cases), [Figures 2 and 3] cervical [Figure 4] and lumbosacral region in 3 patients each [Table 1]. It was astonishing to note that all our patients presented with neurological deficits [Table 2]. Three patients presented with acute meningitis and acute onset paraplegia. History of recurrent meningitis was also positive in two of these cases. Gradually progressing motor deficit was seen in 14 cases. The deficit was in the form of limb weakness and atrophy, with or without gait disturbance. The sensory deficit was seen in 6 cases. Eight patients had bladder/bowel involvement at presentation out of which five were incontinent at the time of presentation. Associated skeletal anomalies were noticed in 5 cases. Scoliosis was the most common finding and was seen in 4 cases, followed by congenital talipes equinovarus in 2 cases. In the majority of the patients (15), sinus ostium was associated with another skin abnormality, the most common of which was abnormal pigmentation. Some patients had a combination of these findings. Dermal sinuses were seen in conjunction with lipomyelomeningocele in 2 patients. MRI was the investigation of choice and was performed in all cases. It revealed the relationship of the dermal sinus to the dural sac and also gave information regarding associated abnormalities in the cord [Table 3]. Epidermoid [Figure 2b] and dermoid tumor [Figure 3b] were seen in 2 and 8 cases respectively. SCM was seen in 6 cases and filum abnormality in 2 cases [Table 4]. Motor deficit (present in 14 cases) stabilized in 7 cases and improved in 6 cases [Table 5]. Two patients developed fresh deficits in the postoperative period, and one of them improved to preoperative status 3 months later. The sensory improvement was seen in 2 cases and sensory deficits stabilized in 4 cases. Neurologic function gradually returned to near normal state postoperatively in 2 of 3 patients who presented with acute paraplegia with incontinence of urine and stool, but there was no improvement in bladder and bowel function in all 3 patients. Of the other 5 incontinent patients, 1 improved, and 4 remained the same.
Figure 1
(a) Spinal dermal sinus tract of lumbar region with lipomyelomeningocele. (b) Magnetic resonance imaging (T1-weighted image) showing the tethered cord due to stalk extending from the sinus to the indradural space along with lipomyelomeningocele
Figure 2
(a) Spinal dermal sinus tract of thoracic region. (b) Magnetic resonance imaging (T2-weighted image) showing intradural epidermoid tumor attached to the thin stalk extending from the sinus
Figure 3
(a) Spinal dermal sinus tract of thoracic region. (b) Magnetic resonance imaging (gadolinium enhanced T1-weighted image) showing intradural dermoid tumor with intramedullary abscess
Figure 4
(a) Spinal dermal sinus tract of cervical region with rudimentary meningocele. (b) Magnetic resonance imaging (T2-weighted image) showing the tethered cord due to stalk extending from the sinus to the indradural space and attach to the bony spur
Table 1
Distribution of spinal DSTs (n=21)
Table 2
Neurological signs and symptoms in patients with dermal sinus (n=21)
Table 3
MRI finding in patients with dermal sinus (n=21)
Table 4
Intraoperative finding in patients with dermal sinus (n=21)
Table 5
Symptom wise surgical outcomes in patients with dermal sinus (n=21)
Overall, 11 patients showed neurological improvement, 9 patients stabilized neurologically while 1 patient deteriorated [Table 6]. Improvement in any of the neuorological parameters viz-motor, sensory or bowel/bladder symptoms was considered to be an improvement.
Table 6
Comparison of surgical outcome with different previous series
A spinal DST consists of a tract lined by stratified squamous epithelium found on or near the midline and is thought to result from the abnormal adhesions (or incomplete disjunction) between the neuroectoderm (destined to form the neural tube) and the cutaneous ectoderm.[4101115] The inward extent of the tract depends upon the extent of adhesions and may vary from deep fascia to the spinal cord. The tract elongates during the development, due to ascent of the cord and may traverse several levels within the epidural space before entering the subarachnoid space. Disorder of the notochord formation with sagittal splitting of the spinal cord and persistence of the dorsal cutaneo-endo-mesenchymal fistula has also been suggested as a cause of dermal sinus formation.[1116] The squamous lining of spinal DST may be encased in dermal and neurological tissue. Within the tract, one may find nerve or ganglion cells or fat, blood vessels, cartilage and meningeal remnants.[14] Spinal DST may be associated with other abnormalities of the ectodermal, mesodermal or neural crest derivatives such as meningomyelocele or lipomeningomyelocele, reflecting a common ontogenic disorder. Nearly, 60% of the DSTs enter the subarachnoid space and 27% are attached to the neural elements of the conus, cauda equina or filum terminale.[151719] The tract may end blindly within the extradural space in 10–20% cases.[617] Sinus tracts can occur anywhere from occiput to sacrum. Different studies showed that cervical area is least involved (<1% cases). Thoracic area is involved in 10% cases, lumbar and lumbosacral area in 40% and 12% patients respectively, sacrum in 23% and sacrococcygeal junction in 13% of cases.[6] In the present study, the cervical region was involved in 14.2% cases, the lumbar region in 52.3% cases, thoracic region in 19% cases and lumbosacral region in 14.2% cases. The higher incidence of DST in cervical area in the present study could be due to selection bias as cervical DST cases are always symptomatic while other authors might include asymptomatic lumbosacral cases.
Dermal sinuses should be distinguished from the more common coccygeal pits. Dermal sinuses are located above the intergluteal cleft and have a cephalically oriented course and are often associated with other pathologies. On the contrary, coccygeal dimples are usually simple blind sinuses with no associated cutaneous abnormalities that lie within intergluteal cleft a few millimeters cranial to the tip of coccyx. They are oriented caudally or straight and are not associated with other intradural pathologies and thus do not warrant further evaluation.[2315] They may rarely have intraspinal extension, so it should be remembered that not all coccygeal pits can be dismissed. Another characteristic that differentiate coccygeal dimple from DST is location. Coccygeal pits are always in midline while DST is not strictly midline and should be investigated with high-quality MRI. If a sacral or coccygeal dimple is associated with other cutaneous abnormalities such as hypertrichosis or soft tissue mass, they should be investigated accordingly.
Dermal sinuses provide a portal of entry for bacterial agents into the intraspinal compartments that can cause meningitis or abscess formation that may be extradural, subdural, and intramedullary or infection of associated tumor. Also, aseptic meningitis can occur by spillage of inclusion tumor contents or other dermal elements into the cerebrospinal fluid.[7919] Therefore, one should have a high level of suspicion for DST and dermoids when encountering any young child presenting with aseptic meningitis. In the study conducted by Jindal and Mahapatra[6] only 1 patient presented with infection out of 26 patients. Ackerman and Menezes[2] also had a low rate (10%) of infectious complications. In the series of Radmanesh et al.,[17] 37.1% had meningitis on admission or had experienced it before while 25.7% had abscess formation. The incidence of infection (meningitis) in our patients was 14.2%, all our infected patients had abscess of which two were intramedullary.
It has been said that nearly all children with spinal DSTs have intact neurological function at birth.[6] However, due to the relatively high rates of associated pathologies such as tethered cord, infection, and inclusion tumors, neurological deterioration becomes more common with increasing age. It has been shown that the chances of developing neurologic deficit are higher in patients who present in older ages.[2] Ackerman and Menezes[2] studied the referral pattern among their patients and noted that patients who were younger than 1-year were more likely to be neurologically intact than older ones, concluding that delay in the diagnosis allows for development of neurologic sequelae. Probably this may be the reason that all of our patients presented with neurological deficits as 90% of our patients were more than 1-year of age which may due to lack of awareness at the primary health care level, which leading to delayed referral. Unfortunately, once a patient develops neurologic deficit, there is a relatively high chance of permanent defect.[56]
Spinal dermal sinuses may be accompanied by other forms of spinal dysraphism such as lipomyelomeningocele and myelomeningocele, reflecting a possible common ontogenic pathway.[17] Gupta et al. showed an association of 11.34% between dermal sinus and other forms of spinal dysraphism.[5] The proposed mechanism for lipomyelomeningocele embryogenesis also includes disorders of disjunction that occurs prematurely in this entity. It is possible that there are some shared molecular pathways responsible for concurrence of these anomalies. Dermal sinuses are occasionally associated with tethered cord, although only 1% of patients with tethered cord have dorsal dermal sinus.[2] In patients with DST, the tract or associated tumor may cause traction on spinal cord resulting in a low-lying conus and tethered cord syndrome.[8] In our study, 13 patients (61.9%) had tethered cord. It is reported that up to 40% of patients with DST can have SCM.[317] Conversely, DSTs are seen in 15–40% of SCM.[19] Among our patients, five had SCM, three with Type 1, and two with Type 2. The incidence of filum terminale abnormalities was described by Jindal and Mahapatra[6] and Radmanesh et al.[17] Jindal and Mahapatra[6] found filum abnormalities in 22% of his patients while Radmanesh et al.[17] found filum terminale abnormality in 40% cases. In the present study, the filum terminale abnormalities were encountered in 5 cases (22%). The term tight filum terminale refers to a set of conditions in which a low-lying conus medullaris is associated with a short thickened filum without evidence of other tethering pathologies.[78] This entity that arises from failed regression of caudal spinal cord during secondary neurulation causes typical signs and symptoms of tethered cord.
Approximately, half of all dermal sinuses are associated with dermoid or epidermoid tumor, usually at the termination of these tracts, but they may be located anywhere between the skin and the neural tube.[261317] Dermal sinuses and dermoid tumors seem to share a common origin.[9] They are believed to result from focal expansion of these ectoderm-derived tracts. However, only approximately 30% of intraspinal dermoid tumors have an associated sinus tract.[7] DSTs are associated more frequently with dermoid tumors (83%) than with epidermoid (13%).[5] In the present study, two of our patients (9.5%) had epidermoid tumors while eight had dermoids (38%) proved by histology.
Postoperative complications were few and easy to manage. Our results indicate that once a patient developed bowel/bladder incontinence, there was about 12.5% chance of improvement in deficit while in patients with sensory or motor deficits; the chance of improvement was 66.6% and 42.8%, respectively [Table 5]. The risk of neurological deterioration was only 3.5%. The patients presenting at later age had more chance of developing deficits. We have also compared the neurological outcomes in the different previous series with our study [Table 6]. In our study, the overall neurological improvement is better than the previous study that may be due to selection bias as all our patients were symptomatic. However, bowel/bladder improvement was seen in only 1 patient due to the delayed presentation.
Since none of imaging modalities can accurately show intraspinal details, all dermal sinuses above the sacrococcygeal region should be explored operatively regardless of neuroimaging findings.[3717] One should have a high index of suspicion for all the dimples above the intergluteal fold, despite a normal examination or neuroradiologic studies. Midline should be carefully examined whenever a child suffers from meningitis, especially when an unusual organism is cultured. Conservative treatment of spinal DST is not recommended. Surgery should be carried out prophylactically in advance of deficits, to maintain normal neurological function.
Spinal DST is an innocuous-appearing spinal dysraphism that may contribute to devastating morbidities if not timely addressed. Although there has been increased awareness about the impotence of dorsal midline cutaneous finding among primary health care physician, there still much more to be done especially in developing country. All patients with spinal DST should be offered aggressive surgical treatment in the form of total excision of sinus tract and correction of spinal malformation, as soon as diagnosed since chances of preserving and/or improving neural function are high (95%).
1. Ackerman LL, Menezes AH, Follett KA. Cervical and thoracic dermal sinus tracts. A case series and review of the literature. Pediatr Neurosurg. 2002. 37: 137-47
3. Elton S, Oakes WJ. Dermal sinus tracts of the spine. Neurosurg Focus. 2001. 10: e4-
4. French BN. The embryology of spinal dysraphism. Clin Neurosurg. 1983. 30: 295-340
5. Gupta DK, Shastank RR, Mahapatra AK. An unusual presentation of lumbosacral dermal sinus with CSF leak and meningitis. A case report and review of the literature. Pediatr Neurosurg. 2005. 41: 98-101
6. Jindal A, Mahapatra AK. Spinal congenital dermal sinus: An experience of 23 cases over 7 years. Neurol India. 2001. 49: 243-6
9. Martínez-Lage JF, Pérez-Espejo MA, Tortosa JG, Ros de San Pedro J, Ruiz-Espejo AM. Hydrocephalus in intraspinal dermoids and dermal sinuses: The spectrum of an uncommon association in children. Childs Nerv Syst. 2006. 22: 698-703
10. McComb JG, Chen TC, Tindall GT, Cooper PR, Barrow DL.editors. Closed neural tube defects. The Practice of Neurosurgery. Baltimore: William and Wilkins; 1996. p. 2753-78
12. Mete M, Umur AS, Duransoy YK, Barutçuoglu M, Umur N, Gurgen SG. Congenital dermal sinus tract of the spine: Experience of 16 patients. J Child Neurol. 2014. 29: 1277-82
13. Morandi X, Mercier P, Fournier HD, Brassier G. Dermal sinus and intramedullary spinal cord abscess. Report of two cases and review of the literature. Childs Nerv Syst. 1999. 15: 202-6
15. Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: Part I: A unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992. 31: 451-80
16. Pang D. Split cord malformation: Part II: Clinical syndrome. Neurosurgery. 1992. 31: 481-500
17. Radmanesh F, Nejat F, El Khashab M. Dermal sinus tract of the spine. Childs Nerv Syst. 2010. 26: 349-57
18. Ramnarayan R, Dominic A, Alapatt J, Buxton N. Congenital spinal dermal sinuses: Poor awareness leads to delayed treatment. Childs Nerv Syst. 2006. 22: 1220-4
19. Tubbs RS, Frykman PK, Harmon CM, Oakes WJ, Wellons JC. An unusual sequelae of an infected persistent dermal sinus tract. Childs Nerv Syst. 2007. 23: 569-71
Background:Since 1976, 10 cases of intradiploic encephaloceles have been reported in the literature. This case is the first report of a spontaneous intradiploic meningoencephalocele of the frontal bone hypothesized to be secondary to distant head trauma.
Case Description:A 60-year-old female with a history of multiple traumatic head injuries as a child presenting with new onset generalized tonic-clonic seizures. Work-up revealed a right frontal epileptic focus. Imaging showed a right frontal intradiploic lesion. The patient underwent surgical resection, which during exploration was found to be an intradiploic encephalocele. She had an uneventful postoperative course with a resolution of seizures.
Conclusions:The authors hypothesize that the rare nature of posttraumatic frontal intradiploic encephaloceles is due to the increased thickness of the frontal bone compared to the parietal bone.
In 1976, Kosnik et al. published the first report of an intradiploic encephalocele.[6] The patient was a 57-year-old man who presented with generalized seizures and expressive aphasia.[6] Work-up revealed a left parietal lytic bone lesion on skull films, and an electroencephalogram (EEG) showed moderate slowing at 4 Hz with spike activity in the region underlying the skull lesion.[6] The patient underwent surgical resection of the encephalocele with a postoperative resolution of seizures.[6] Since this time, nine additional intradiploic encephaloceles have been reported in the literature. Eight of these cases were encephaloceles of the parietal bone, and the ninth was an iatrogenic encephalocele of the frontal bone.[134789121315] We report the first spontaneous intradiploic meningoencephalocele of the frontal bone, which we hypothesize to be secondary to trauma from the distant past.
A 60-year-old female has a history of multiple head traumas as a young adult, without documented skull fracture, resulting from multiple falls while horseback riding. Her past history includes type two diabetes controlled with lifestyle modification and breast cancer in 2002 treated with surgical resection (2002) and chemoradiation (2003). She first presented with generalized tonic-clonic seizure. Her physical exam was notable for a palpable hard protuberance on her right frontal calvarium. An EEG showed intermittent low-amplitude polymorphic delta slowing over the right anterior head region, without clear epileptiform discharges, and she was treated with levetiracetam for seizure prophylaxis. Her neurological exam remained normal, and she did not have any subsequent seizures.
Imaging studies of her head were performed. The computed tomography (CT) of her head demonstrated a 3.7 cm × 3.4 cm × 1.5 cm lytic lesion of the right frontal calvarial bone with the erosion of the inner table [Figure 1]. The magnetic resonance imaging (MRI) of her brain suggested herniation of the right frontal lobe parenchyma into the bony defect [Figure 2].
Figure 1
(a) Axial computed tomography head demonstrating erosion of the inner table of the right frontal bone. (b) Coronal computed tomography head demonstrating erosion of the inner table of the right frontal bone
Figure 2
(a) Axial T1-weighted magnetic resonance imaging with heterogeneous hypointensity of the right frontal lobe lesion. (b) Axial T2-weighted magnetic resonance imaging with heterogeneous hyperintensity of right frontal lobe lesion. (c) Coronal T2-weighted magnetic resonance imaging with heterogeneous hyperintensity of right frontal lobe lesion
Operation
Due to the history of seizure activity and the expansile nature of the bony mass, the patient underwent surgical resection. A right frontotemporal craniotomy for resection of the right frontal calvarial lesion was performed. Upon inspection of the skull prior to the craniotomy, the lesion was evident by marked cystic thinning of the frontal bone. As the bone flap was elevated, brain tissue was noted to have herniated into the calvarial lesion [ Figure 3a ]. Bipolar electrocautery was used to coagulate and divide the herniated portion of the brain from the right frontal lobe, and the specimen was sent for frozen and permanent pathology. The bone flap was inspected on the surgical table [ Figure 3b ], and the inner table was removed prior to the replacement of the bone flap and closure.
Figure 3
(a) Intraoperative photograph of meningoencephalocele herniating through the inner calvarial table. (b) Intraoperative photograph of fontal bone lesion
Pathology
Frozen and permanent sections were taken of both the herniated brain tissue and bone lesion. The frozen sections demonstrated no definitive pathology with herniated cerebral cortex as well as a bone with fibrous tissue. The permanent sections, stained with hematoxylin and eosin [ Figure 4 ], confirmed the frozen findings and also demonstrated gliosis of the cerebral cortex with thickening of the leptomeninges within the defect in the skull.
Figure 4
(a) Cerebral cortex with a bone, that is, intermingled with arachnoidal cells; H and E (scale bar = 100 μm). (b) Gliotic cerebral cortex with thickened leptomeninges; H and E (scale bar = 100 μm)
Herniation of cerebral contents into the intradiploic space is an uncommon clinical scenario. Case reports have demonstrated multiple different etiologies including meningoencephaloceles, as in the above case, as well as giant arachnoid granulations, epidermoid cysts, and arachnoid cysts.[23514] A cephalocele is a herniation of intracranial contents through a bony or dural defect.[7] They are classified both by location (e.g., frontal, parietal, and occipital) and the herniated contents: meninges (meningeal) or meninges and parenchyma (meningoencephaloceles). Most commonly, cephaloceles are congenital defects occurring in the midline secondary to improper closure of the neural tube. Acquired cephaloceles occurring during adulthood have been attributed many etiologies, including: Infection, trauma, surgery, and tumors.[37] When there is no easily identifiable cause, they are known as spontaneous cephaloceles, and most commonly occur in the cranial sutures.[3] In this case, the spontaneous meningoencephalocele is hypothesized to have occurred secondary to a skull fracture in the distant past, although no clear documentation of such is found.
Intradiploic meningoencephaloceles are the result of a defect of the inner table of the calvarium and subsequent herniation of the meninges and cerebral parenchyma into the intradiploic space. Of the ten previously reported cases, nine presented within the parietal bone, and the tenth occurred within the frontal bone.[134789121315] It is important to note that the etiology of the single case within the frontal bone was iatrogenic, occurring after an accidental tear of the dura during a craniosynostosis repair.[9] The presentation of intradiploic meningoencephaloceles tends to be associated with the location of the lesion and includes a headache, focal weakness, and seizures.[38]
Imaging studies aid the diagnosis of intradiploic encephaloceles. On CT, there is an erosion of the inner calvarial table, and on MRI the herniated parenchyma will appear hypointense on T1-weighted images and hyperintense on T2-weighted images.[3] Diagnosis is confirmed with a tissue sample demonstrating cerebral parenchyma, often with evidence of gliosis, and no evidence of other pathologies.[3] The clinical presentation, radiographic, and pathologic findings of the patient in this report are consistent with an intradiploic meningoencephalocele. In fact many of the findings including, the patient's age, presenting symptoms, and clinical course are very similar to report by Kosnik et al. from 1976.[6] However, two distinctions make this case notable. First, the patient's history of multiple traumatic head injuries and second the frontal location of the meningoencephalocele.
Of the 10 previously reported cases of intradiploic encephaloceles, only one was associated with head trauma. This case, reported by Patil and Etemadrezaie occurred in a 61-year-old male who hit had hit his head on a garage door approximately 1-year prior to presenting with a persistent lump on his head and no neurologic symptoms.[12] In this report, the authors compared the intradiploic meningoencephalocele to other posttraumatic intradiploic cystic lesions, including arachnoidal cysts, leptomeningeal cysts, and cerebrospinal fluid (CSF) fistula.[12] The authors hypothesized that posttraumatic intradiploic meningoencephaloceles are a rare adult variants of growing skull fractures, where low-velocity blunt trauma only fractures the inner table, as opposed to a growing skull fracture in which both the inner and outer tables are disrupted.[1112] The fracture of the inner table results in a dural tear; as the skull recoils, a negative pressure draws the underlying arachnoid and parenchyma into the intradiploic space.[12] In addition, posttraumatic intradiploic encephaloceles and growing skull fractures differ in the extent of damage to the underlying parenchyma. Growing skull fractures usually result in secondary and often severe damage to the underlying parenchyma due to the underlying mechanism of rapid brain growth and CSF pulsation in infants.[11] However, in the previously reported posttraumatic intradiploic encephalocele as well as in the present case there was minimal damage to the underlying parenchyma.[12] This is likely due to the head injuries occurring after the period of rapid brain growth signified by the closure of the cranial sutures, and thus limiting the mechanism of secondary damage to the underlying parenchyma.
The frontal location is an additional distinction in this patient's meningoencephalocele. This case is the first reported spontaneous intradiploic encephalocele of the frontal bone. It is unclear why frontal intradiploic encephaloceles are less common than those of the parietal bone. A possible explanation is the difference in the average thickness of the two bones. The parietal bone is on average thinner than the frontal bone,[10] and is, therefore, likely to be more susceptible to developing posttraumatic intradiploic encephaloceles. However, this does not explain the increased frequency of spontaneous parietal intradiploic encephaloceles. Given that the trauma in posttraumatic intradiploic encephaloceles may occur 1 or more years prior to presentation without neurological deficit, many seemingly spontaneous intradiploic encephaloceles may be posttraumatic with the traumatic event not recalled or believed to be relevant by the patient. If trauma is the underlying cause of many spontaneous intradiploic encephaloceles, then this hypothesis would explain the increased incidence of intradiploic encephaloceles in the parietal bone as compared to the frontal bone.
Posttraumatic intradiploic meningoencephaloceles remain a rare clinical entity. They are most likely a variant form of growing skull fractures and may be the underlying etiology of many spontaneous intradiploic encephaloceles. This case represents the first reported spontaneous intradiploic meningoencephalocele of the frontal bone which is believed to have occurred secondary to head trauma in the distant past.
2. Chan WC, Lai V, Wong YC, Poon WL. Focal brain herniation into giant arachnoid granulation: A rare occurrence. Eur J Radiol Extra. 2011. 78: e111-3
3. Dobrin N, Mihaela B, Cost B, Tudorache C, Chiriac A, Poeat I. Acquired parietal intradiploic encephalocele. Case report and review of the literature. Romanian Neurosurg. 2011. p. 18-
4. Froelich S, Botelho C, Abu Eid M, Kehrli P, Dietemann JL, Maitrot D. Encéphalocèle intra-diploïque de l’adulte. Neurochirurgie. 2006. 52: 551-4
14. Peters SA, Frombach E, Heyer CM. Giant arachnoid granulation: Differential diagnosis of acute headache. Australas Radiol. 2007. 51: B18-20
15. Tsuboi Y, Hayashi N, Noguchi K, Kurimoto M, Nagai S, Endo S. Parietal intradiploic encephalocele - Case report. Neurol Med Chir (Tokyo). 2007. 47: 240-1
Department of Pediatric Neurosurgery, Dana Children's Hospital, Tel-Aviv Medical Center, Tel-Aviv University, Tel-Aviv, Israel
Department of Ophthalmology, Neuro-ophthalmology Unit, Tel-Aviv Medical Center, Tel-Aviv University, Tel-Aviv, Israel
Correspondence Address: Jonathan Roth Department of Ophthalmology, Neuro-ophthalmology Unit, Tel-Aviv Medical Center, Tel-Aviv University, Tel-Aviv, Israel
How to cite this article: Roth J, Constantini S, Kesler A. Over-drainage and persistent shunt-dependency in patients with idiopathic intracranial hypertension treated with shunts and bariatric surgery. Surg Neurol Int 08-Dec-2015;6:
Background:Idiopathic intracranial hypertension (IIH) may lead to visual impairment. Shunt surgery is indicated for refractory IIH-related symptoms that persist despite medical treatment, or those presenting with significant visual decline. Obesity is a risk factor for IIH; a reduction in weight has been shown to improve papilledema. Bariatric surgery (BS) has been suggested for treating IIH associated with morbid obesity. In this study, we describe a high rate of over-drainage (OD) seen in patients following shunts and BS.
Methods:The study cohort includes 13 patients with IIH that underwent shunt surgery for treatment of the IIH-related symptoms. Six patients underwent BS in addition to the shunt surgery (but not concomitantly). Seven patients had only shunt surgeries with no BS. Data were collected retrospectively.
Results:BS effectively led to weight reduction (body mass index decreasing from 43 ± 4 to 28 ± 5). Patients undergoing BS had 1–6 (2.5 ± 1.9) shunt revisions for OD following BS, as opposed to 0–3 (1.4 ± 1.1) revisions prior to BS over similar time spans (statistically insignificant difference), and 0–6 (1.6 ± 2.5) revisions among the non-BS patients over a longer time span (statistically insignificant difference). Two patients in the BS group underwent shunt externalization and closure; however, they proved to be shunt-dependent.
Conclusions:Patients with IIH that undergo shunt surgery and BS (not concomitantly) may suffer from OD symptoms, necessitating multiple shunt revisions, and valve upgrades. Despite BS being a valid primary treatment for some patients with IIH, among shunted patients, BS may not lead to resolution of IIH-related symptoms and patients may remain shunt-dependent.
Idiopathic intracranial hypertension (IIH), also known as pseudotumor cerebri, is a disease of unknown origin, affecting primarily young women, often with morbid obesity. Major clinical presentations are headaches and transient visual obscurations. Treatment typically consists of medications (such as acetazolamide or Topamax) that decrease cerebrospinal fluid (CSF) production. Patients with refractory headaches and/or severe visual decline are often treated by various surgical options, including optic nerve sheath fenestration (ONSF) and CSF diversion procedures such as lumboperitoneal shunt (LPS) or ventriculoperitoneal shunt (VPS). Despite the proven positive effect of CSF diversion procedures on visual outcome, many patients continue to suffer from chronic headaches, and often undergo multiple shunt revisions (treating symptoms of over and under-drainage) or multiple episodes of intracranial pressure monitoring (ICPm) to try and correlate the symptoms to ICP.[25]
Recently, bariatric surgery (BS) has gained popularity, and has proven to be effective in weight reduction, as well as improvement of various obesity-related morbidities such as diabetes, ischemic heart disease, and general wellbeing.[162326]
Previous reports among patients with IIH and obesity have shown that BS may lead to major symptom relief and visual improvement.[3511131820272829] Despite these encouraging publications, there is a scarcity of information on the effect of BS on obese patients that had prior CSF diversion surgeries.[15]
This study focuses on patients diagnosed with IIH and treated with a shunt, who underwent BS at a later stage, or those treated primarily with BS, followed at a later stage with a shunt.
Following an Institutional Review Board approval, data were collected retrospectively. Patient approval was waived. Collected data included: Demographics, surgical history (relating to ONSF, shunt surgeries, ICPm, and BS), and clinical course. Clinical data included degree of headache (none [minimal impact], existing [with no major impact on daily life], severe [with significant impact on daily life]), and data relating to patients’ weight.
Ophthalmological variables included visual acuity (VA), optic disc appearance (ODA), and visual fields (VFs).
Between 2002 and June 2014, 14 patients, all females, underwent CSF diversion procedures (LPS or VPS) for the treatment of IIH. Of these, 7 have undergone both BS and shunt procedures. However, 1 of the patients underwent BS several years before diagnosing IIH, undergoing a gastric banding procedure 9 years prior to an LPS while had no symptoms of IIH. The banding sleeve in this patient was removed 1 year later due to abdominal complications. One patient from the non-BS group was lost to follow-up after the shunt surgery, and thus was not included in this study. Thus, for practical reasons, we divided the patients into the following categories: Those that underwent both BS and a shunt (6 patients), and those that did not undergo BS (7 patients, including the patient that underwent a band removal 9 years prior to the shunt surgery).
Shunt revisions were categorized as primary shunt insertions, revisions due to shunt infections, revisions treating under-drainage symptoms (originating from a restrictive valve), revisions treating over-drainage (OD) symptoms (originating from a permissive valve), and technical revisions (for treating disconnections, catheter migrations, and valve rotations).
Data were summarized in an excel file, and basic statistical analysis (mean ± standard deviation) was performed for the numerical data. Comparison of number of surgeries done in the various groups was done using an unpaired Student's t-test.
Of the 6 patients in the BS group, 5 had an LPS 3–70 months (43 ± 26) prior to the BS, and 1 had a VPS 2 years following BS. Ages at shunt surgeries were 21–31 years (27 ± 4). Two patients underwent ONSF prior to the shunt. Prior to BS, patients underwent an average of 3–11 shunt revisions (6.2 ± 3.3). The indication for BS was for weight reduction (and not control of IIH). The indication for shunt (in both the BS and non-BS groups) was the treatment of refractory IIH despite maximal medication treatment, accompanied with a visual decline that necessitated immediate pressure reduction.
Four patients underwent sleeve gastrectomy, and 2 underwent gastric banding (see an overview on BS techniques and efficacy by DeMaria, 2007).[ 10 ] At the time of the shunt surgeries, patients weighed 95–118 kg (109 ± 9), and had a body mass index (BMI) of 37–47 (43 ± 4). Following BS, patients lost 5–54 kg (37 ± 17) over a period of 10–84 months (39 ± 25), and the BMI reduced in all patients (from 43 ± 4 to 28 ± 5). Following BS, patients underwent 1–14 (5.7 ± 5) shunt related surgeries.
Nonbariatric surgery group
Of the 7 patients in the non-BS group, 6 underwent LPS and 1 VPS. Ages at primary surgery were 12–43 years (26 ± 10). Two patients underwent ONSF prior to the shunt surgery. At the time of shunt surgeries, patients weighed 60–120 kg (92 ± 23), and had a BMI of 26–43 (35 ± 7). At last follow-up, patients weighed 56–126 kg (88 ± 21), and had a BMI of 22–45 (33 ± 8).
Clinical course and shunt revisions
Follow-up duration after the first shunt procedure was 23–181 (86 ± 47) months for the entire group (39–116 (75 ± 25) in the BS group, and 23–181 (95 ± 61) in the non-BS group). Follow-up period following the BS procedure was 10–84 (39 ± 25) months.
All the BS patients underwent multiple shunt revisions due to various indications (as stated in the methods section). The exact surgical course was available for 11 patients (the 6 BS patients, and 5 of 7 non-BS patients). Only 2 patients underwent 1 revision each for under-drainage. We focused on patients undergoing revisions for OD and technical reasons [ Table 1 ]. Regarding OD indications, the BS group had 1–3 (1.4 ± 1.1) revisions prior to the BS, but 1–6 (2.5 ± 1.9) revisions for after BS. The non-BS group underwent 0–6 (1.6 ± 2.5) revisions for OD indications. The difference in number of revisions due to OD did not reach statistical significance between the various patient groups. Following OD, valves were upgraded, changed to dual switch valve (DSV), or added an assist device. Current shunt valves are summarized in [ Table 2 ].
Table 1
Number of shunt revisions
Table 2
Current valve status
Regarding technical indications, prior to BS, patients underwent 0–5 (2.4 ± 1.8) revisions, as opposed to 0–5 (1.5 ± 1.9) following BS, and 0–4 (1.4 ± 1.9) revisions in the non-BS group.
In 2 of the 5 patients that underwent BS following LPS, the LPS was externalized to evaluate the need for any CSF diversion. These patients lost 47 and 42 kg and had clear OD symptoms. Both patients had the shunt externalized and elevated gradually, and developed severe under-drainage symptoms over a course of hours, which improved once the height of the bag was lowered. Both patients improved with shunt revisions.
At last follow-up, 2 patients in the BS group, and 4 in the non-BS group still complain of headache.
Visual exams
Preshunting visual evaluation was available for 12 of 13 patients (24 eyes). VA was intact or only mildly compromised in 22 eyes preshunting and in 19 eyes postshunting. Overall, VA deteriorated in 3 eyes. VFs were intact or with minor defects in 17 eyes preshunting, and in 20 postshunting. Overall, VF deteriorated in 4 eyes and improved in 7. ODA was normal in only 1 eye before surgery but was normal in 15 following shunt surgery. Overall, the ODA improved in 12 eyes and deteriorated in 7.
When comparing the BS and the non-BS groups, both had similar improvement and deterioration numbers in both VA and VF. However, ODA normalized in 6 eyes in the BS as opposed to 4 in the non-BS group. Of the 6 patients with remaining headaches, 1 had no available preoperative visual evaluation. The remaining 5 had stable intact VA (8 eyes), deteriorated VA (2 eyes), stable VF (5 eyes), deteriorated VF (2 eyes), and improved VF (3 eyes). ODA was stable (1 eye), deteriorated (4 eyes), and improved (5 eyes) at last follow-up.
This is the largest report on the effect of BS on IIH patients that have a functioning shunt. The study points out two main issues:
OD is common following shunt surgeries for IIH. However, OD is more common in patients with a shunt and BS
While BS and the subsequent weight loss can potentially cure IIH in nonshunted patients; patients with a shunt already in place may remain shunt-dependent even after BS. Two shunted patients that underwent BS, developed OD, and were subsequently challenged for shunt dependency. Both proved to be shunt-dependent.
IIH is a poorly understood disease, which typically affects young, obese women. The association of IIH to overweight has been repeatedly described, although the mechanism is poorly understood. Between 64% and 70% of IIH patients are obese[3] High BMI is a risk factor for IIH,[9] and a poor prognostic factor once IIH has been diagnosed.[3] Several mechanisms have been suggested to correlate between overweight and IIH, including mechanical reasons such as increased intra-abdominal and intrathoracic pressures leading to reduced intracranial venous drainage, and hormonal reasons such as elevated leptin and estrogen.[259] Other contributing factors may include hypoventilation and sleep apnea associated with obesity, leading to hypercarbia and elevated ICP,[35] or occult cerebral sinus thrombosis affecting cerebral venous drainage.[3]
Treatment of IIH includes diet and medications such as acetazolamide and Topamax. However, in refractory IIH causing incapacitating headaches or continuous visual decline, surgical alternatives are applied, such as ONSF and CSF diversion surgeries.[17] Currently, there are no clear indications for surgical treatment or technique; decisions are based on each center's experience.[4] In general, it is accepted that following both ONSF and CSF diversions, headache remains a common complaint over the years in about 40–60% of patients.[141719212530] In additional, both LPS and VPS have a high rate of malfunction, necessitating shunt revisions, mostly due to OD or blockage.[2125303133] Many valves have been utilized to treat and prevent OD; our personal experience has been to place horizontal-vertical valves (such as the DSV by Miethke), sometimes coupled with a gravitational component (shunt assist [SA], by Miethke).[32] DSV includes two valves that toggle between them depending on the patient's position. At the upright position, the higher pressure valve functions while at the horizontal position, the lower pressure valve functions. This mechanism reduces OD occurrence compared with single valve systems.[32]
Weight loss has shown positive effects on IIH-related symptoms, especially when meaningful weight reduction is achieved.[316] BS in its various techniques has been repeatedly shown to cause not only subjective improvement in symptoms such as headaches and visual decline but also objective improvement in visual tests[513] and reduction in lumbar puncture pressure.[2429] These improvements in IIH-related symptoms, together with other health related advantages of weight reduction, have made BS a valid and even a preferred alternative for IIH treatments,[3511131820272829] and is increasingly utilized for IIH.[7] Despite these encouraging results, shunt dependency has been reported following BS, although rare.[8]
The main finding of the current series highlights the association of BS with OD symptoms in a small group of patients that had undergone both BS and shunt surgery (not concurrently). This concept of BS-induced shunt OD has been described in a previously published case report of a patient with an LPS that underwent BS and developed secondary Chiari.[15] Symptoms and radiological findings responded to a valve upgrade.
The mechanisms by which BS may induce OD are unclear, but may include:
Cure or improvement of IIH by weight reduction, thus making the CSF diversion redundant. It has been suggested by several authors that in this context, when significant long-term weight loss is anticipated based on a planned BS, LPS may be a valid interim solution for IIH despite the “short life expectancy of these shunts”[2025]
Reduced abdominal pressure secondary to weight loss may increase the pressure gradient across the shunt valve and lead to OD. We speculate that this is a major factor, and thus have continued to upgrade the valve system (eventually achieving good clinical outcome)
Reduced abdominal pressure leads to a reduction of pressure in the inferior vena cava. This may increase venous drainage from the epidural venous (Batson) plexuses. In a similar fashion, it has been suggested that increased drainage from these plexuses may be the basis for spontaneous intracranial hypotension[12]
BS leading to significant weight loss improves sleep study parameters and reduces obstructive sleep apnea (OSA).[22] OSA is thought to contribute to elevated ICP levels.
When relating to shunt revisions due to technical reasons (tubing disconnections, catheter migrations, and valve rotations), the rate prior to BS was higher than after. This may pertain to obesity-related factors that lead to shunt migration and valve rotations. However, the numbers are too small to draw any concrete conclusions.
Another important observation in this small patient group was the incidence of shunt dependency even after BS with effective weight reduction. In 2 patients, following several valve upgrades, we suspected that the BS had cured the IIH and actually made CSF diversion unneeded. Thus, both patients underwent externalization of the shunt. Both patients proved to be shunt-dependent with a low compliance needing effective CSF drainage within hours. We suspect that a potential explanation may be that patients with shunts have a more severe form of IIH as compared to the nonshunted patients. The shunted patients have more severe presenting symptoms of headaches or visual decline and are more prone to be refractory to medical treatments. Thus, weight loss may not be sufficient to cure their IIH.
Our current policy in the treatment of IIH is medical treatment combined with weight loss programs (including BS). When there is deterioration in visual function despite maximum treatment, we advocate CSF diversion (preferably LPS). We advocate using a restricted valve (such as the DSV) and avoid using programmable valves (as it is difficult to palpate the exact location of the valve and assure a correct pressure programming). Despite the disappointing results of BS on IIH-related headaches in this group of previously shunted patients, BS has an important role on the general wellbeing of IIH patients suffering from morbid obesity, and we encourage patients to pursue it. We acknowledge and instruct our patients that OD symptoms may arise, and that a need for valve upgrades may arise once weight loss is achieved. If these symptoms occur, we upgrade the DSV to higher pressures and add an SA, or convert the LPS to VPS. It is accepted that VPS tend to have less malfunctions (especially obstructions) and less chiari than LPS; however, OD rates seem to be generally similar.[1930] The reason for less shunt obstructions in VPS compared to LPS is unknown, but may be associated with the larger tube diameter and lumen compared to LPS. VPS may be more adequate for programmable shunts, as they are adjacent to the skull and safer to program.
This study has several limitations. First, the small retrospective group limits the ability to generalize our results and even perform meaningful statistics. Second, data concerning indication for shunt revision and the exact valve details were missing for 2 of 7 non-BS patients (they were previously treated elsewhere). Third, the two groups (BS and non-BS) may not represent the same IIH severity, for instance because of significantly different BMI values. Fourth, it is not absolute that BS was the trigger to the OD symptoms, as these symptoms occurred prior to BS (although at a lower rate), and also in the non-BS group. However, the rate of OD following BS (100%), makes BS a probable contributing factor in the OD occurrence.
BS has an important role treating morbidly obese patients with IIH. In some patients, this may result in improvement of IIH-related symptoms. However, in patients that underwent a previous shunt treatment, or in patients with prior BS that undergo a later shunt placement, BS may provoke OD symptoms, leading to future shunt related surgeries. In additional, these shunted patients may still be shunt-dependent despite undergoing BS and significantly reducing their BMI.
1. Alosco ML, Galioto R, Spitznagel MB, Strain G, Devlin M, Cohen R. Cognitive function after bariatric surgery: Evidence for improvement 3 years after surgery. Am J Surg. 2014. 207: 870-6
3. Banik R. Obesity and the role of nonsurgical and surgical weight reduction in idiopathic intracranial hypertension. Int Ophthalmol Clin. 2014. 54: 27-41
4. Brazis PW. Clinical review: The surgical treatment of idiopathic pseudotumour cerebri (idiopathic intracranial hypertension). Cephalalgia. 2008. 28: 1361-73
5. Chandra V, Dutta S, Albanese CT, Shepard E, Farrales-Nguyen S, Morton J. Clinical resolution of severely symptomatic pseudotumor cerebri after gastric bypass in an adolescent. Surg Obes Relat Dis. 2007. 3: 198-200
6. Courcoulas AP, Christian NJ, Belle SH, Berk PD, Flum DR, Garcia L. Weight change and health outcomes at 3 years after bariatric surgery among individuals with severe obesity. JAMA. 2013. 310: 2416-25
7. Curry WT, Butler WE, Barker FG. Rapidly rising incidence of cerebrospinal fluid shunting procedures for idiopathic intracranial hypertension in the United States, 1988-2002. Neurosurgery. 2005. 57: 97-108
8. D’Alessandris QG, Montano N, Bianchi F, Doglietto F, Fernandez E, Pallini R. Persistence of primary empty sella syndrome despite obesity surgery: Report of two unusual cases. Br J Neurosurg. 2012. 26: 875-6
9. Daniels AB, Liu GT, Volpe NJ, Galetta SL, Moster ML, Newman NJ. Profiles of obesity, weight gain, and quality of life in idiopathic intracranial hypertension (pseudotumor cerebri). Am J Ophthalmol. 2007. 143: 635-41
10. DeMaria EJ. Bariatric surgery for morbid obesity. N Engl J Med. 2007. 356: 2176-83
11. Egan RJ, Meredith HE, Coulston JE, Bennetto L, Morgan JD, Norton SA. The effects of laparoscopic adjustable gastric banding on idiopathic intracranial hypertension. Obes Surg. 2011. 21: 161-6
12. Franzini A, Messina G, Nazzi V, Mea E, Leone M, Chiapparini L. Spontaneous intracranial hypotension syndrome: A novel speculative physiopathological hypothesis and a novel patch method in a series of 28 consecutive patients. J Neurosurg. 2010. 112: 300-6
13. Fridley J, Foroozan R, Sherman V, Brandt ML, Yoshor D. Bariatric surgery for the treatment of idiopathic intracranial hypertension. J Neurosurg. 2011. 114: 34-9
14. Friedman DI, Rausch EA. Headache diagnoses in patients with treated idiopathic intracranial hypertension. Neurology. 2002. 58: 1551-3
15. Graber JJ, Racela R, Henry K. Cerebellar tonsillar herniation after weight loss in a patient with idiopathic intracranial hypertension. Headache. 2010. 50: 146-8
16. Johnson LN, Krohel GB, Madsen RW, March GA. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri). Ophthalmology. 1998. 105: 2313-7
17. Lai LT, Danesh-Meyer HV, Kaye AH. Visual outcomes and headache following interventions for idiopathic intracranial hypertension. J Clin Neurosci. 2014. 21: 1670-8
18. Leslie DB, Kellogg TA, Boutelle KN, Barnett SJ, Schwarzenberg SJ, Harrison AR. Preserved vision without growth retardation after laparoscopic Roux-en-Y gastric bypass in a morbidly obese child with pseudotumor cerebri: 36-month follow-up. J Pediatr Surg. 2008. 43: e27-30
19. McGirt MJ, Woodworth G, Thomas G, Miller N, Williams M, Rigamonti D. Cerebrospinal fluid shunt placement for pseudotumor cerebri-associated intractable headache: Predictors of treatment response and an analysis of long-term outcomes. J Neurosurg. 2004. 101: 627-32
20. Nadkarni T, Rekate HL, Wallace D. Resolution of pseudotumor cerebri after bariatric surgery for related obesity. Case report. J Neurosurg. 2004. 101: 878-80
21. Niotakis G, Grigoratos D, Chandler C, Morrison D, Lim M. CSF diversion in refractory idiopathic intracranial hypertension: Single-centre experience and review of efficacy. Childs Nerv Syst. 2013. 29: 263-7
22. Ravesloot MJ, Hilgevoord AA, van Wagensveld BA, de Vries N. Assessment of the effect of bariatric surgery on obstructive sleep apnea at two postoperative intervals. Obes Surg. 2014. 24: 22-31
23. Reddy RC, Baptist AP, Fan Z, Carlin AM, Birkmeyer NJ. The effects of bariatric surgery on asthma severity. Obes Surg. 2011. 21: 200-6
24. Sinclair AJ, Burdon MA, Nightingale PG, Ball AK, Good P, Matthews TD. Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: Prospective cohort study. BMJ. 2010. 341: c2701-
25. Sinclair AJ, Kuruvath S, Sen D, Nightingale PG, Burdon MA, Flint G. Is cerebrospinal fluid shunting in idiopathic intracranial hypertension worthwhile?. A 10-year review. Cephalalgia. 2011. 31: 1627-33
26. Sjöström L, Peltonen M, Jacobson P, Ahlin S, Andersson-Assarsson J, Anveden Å. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA. 2014. 311: 2297-304
27. Soto FC, Antozzi P, Szomstein S, Cho MY, Zundel N, Locatelli E. Indication for emergent gastric bypass in a patient with severe idiopathic intracranial hypertension: Case report and review of the literature. Surg Obes Relat Dis. 2005. 1: 503-5
28. Stangherlin P, Ledeghen S, Scordidis V, Rubay R. Benign intracranial hypertension with recurrent spontaneous cerebrospinal fluid rhinorrhoea treated by laparoscopic gastric banding. Acta Chir Belg. 2008. 108: 616-8
29. Sugerman HJ, Felton WL, Sismanis A, Kellum JM, DeMaria EJ, Sugerman EL. Gastric surgery for pseudotumor cerebri associated with severe obesity. Ann Surg. 1999. 229: 634-40
30. Tarnaris A, Toma AK, Watkins LD, Kitchen ND. Is there a difference in outcomes of patients with idiopathic intracranial hypertension with the choice of cerebrospinal fluid diversion site: A single centre experience. Clin Neurol Neurosurg. 2011. 113: 477-9
31. Toma AK, Dherijha M, Kitchen ND, Watkins LD. Use of lumboperitoneal shunts with the Strata NSC valve: A single-center experience. J Neurosurg. 2010. 113: 1304-8
32. Udayakumaran S, Roth J, Kesler A, Constantini S. Miethke DualSwitch Valve in lumboperitoneal shunts. Acta Neurochir (Wien). 2010. 152: 1793-800
33. Yadav YR, Parihar V, Agarwal M, Bhatele PR, Saxena N. Lumbar peritoneal shunt in idiopathic intracranial hypertension. Turk Neurosurg. 2012. 22: 21-6
Department of Neurosurgery, University Hospital Essen, Essen, Germany
Institute of Diagostic and Interventional Radiology and Neuroradiology, University Hospital Essen, Essen, Germany
Department of Neurosurgery, Klinikum Nordstadt Hannover, Hannover, Germany
Correspondence Address: Neriman Özkan Department of Neurosurgery, University Hospital Essen, Essen, Germany Department of Neurosurgery, Klinikum Nordstadt Hannover, Hannover, Germany
How to cite this article: Neriman Özkan, Jabbarli R, Wrede KH, Sariaslan Z, Stein KP, Dammann P, Ringelstein A, Sure U, Sandalcioglu EI. Surgical management of intradural spinal cord tumors in children and young adults: A single-center experience with 50 patients. Surg Neurol Int 08-Dec-2015;6:
Background:Intradural spinal cord tumors (IDSCTs) in children and young adults are rare diseases. This present study is aimed to demonstrate our experience with a large series of children and young adults with IDSCT.
Methods:A total of 50 patients aged
Results:Mean age was 10.3 years (range 6 months–19 years). IDSCT surgery was performed in 44 patients (88%). A common first symptom in patients with EMSCT was neck and back pain (41%), whereas monoparesis of arms (43%) were often seen in patients with IMSCT. The main duration of the symptoms was longer in patients with IMSCT. The postoperative functional outcome was generally comparable to the preoperative functional condition, while better for EMSCT (P P
Conclusion:Due to the mostly mild impact of the surgery on the functional outcome, the surgical treatment of IDSCT in children and young patients can be uniquely advocated.
Spinal tumors are rare and compromise approximately 5–10% of all tumors of the central nervous system in children and young adults.[32] The annual incidence of these tumors varies between 0.9 and 2.6/100,000.[24]
Since the first successful removal of an intradural extramedullary fibromyxoma by Gowers and Horsley in 1888,[21] remarkable progress has been achieved in spinal cord tumor surgery. Advancements in diagnostics, surgical techniques equipment, and oncological treatment improved the outcome of these challenging tumors, in particular, with intramedullary involvement.[152439] The development of modern neuroimaging procedures for surgical planning, as well as innovative technologies for intraoperative visualization and tumor resection, represent substantial developments which have contributed to the safety and efficacy of spinal cord surgery.[37121720344344]
The age-related peculiarities of the surgical treatment of spinal cord tumors in children and young adults lie in the body growth and different biological features of neoplastic lesions. However, there are still no specific guidelines or recommendations for the treatment of spinal cord tumors in children and young patients.[22]
Against this background, we present a large series of children and young adults with intradural spinal cord tumors (IDSCTs) treated at our department during a 20-year period and aimed to derive specific treatment recommendations for pediatric patients upon a single-center experience.
Data of 90 pediatric patients and young adults (younger than 20 years) with the diagnosis of a spinal cord lesion, who were treated at our Neurosurgical Department between January 1990 and December 2010, were retrospectively collected. Children and young adults with extradural spinal cord tumors and spinal abscesses, as well as intradural vascular lesions such as cavernous hemangiomas, arteriovenous fistulas, and arteriovenous malformations, were excluded from this study. Thus, a total of 50 patients fulfilling the inclusion criteria of IDSCTs were identified and enrolled into the study. The study was conducted in accordance with the Declaration of Helsinki and the Guideline for Good Clinical Practice. The study protocol was approved by the local ethic committee of the University of Duisburg-Essen, Germany.
Data management
The data on demographic, clinical, and radiological characteristics of the patients, intraoperative findings, and complications, as well as the parameters of the postoperative course were retrospectively collected. The neurological status was assessed using the Frankel score.[ 19 ] According to this score, the outcome was classified as poor (A + B), fair (C), and good (D + E).
All patients underwent the clinical examination pre- and post-operatively, as well as 3 months after the surgery. Radiological examination using magnetic resonance imaging (MRI) before and 3 months after surgery was also performed in all cases. Furthermore, depending on the histopathology and clinical status, patients underwent later clinical and MRI-controls (at 6 months and yearly) in certain cases. Therefore, the average follow-up period was 3.5 ± 3.2 years (range 3 months–10 years).
Surgical treatment was performed under standard microsurgical conditions and intraoperative electrophysiological monitoring (with obligatory use of sensory evoked potentials, as well as increasing the use of motor-evoked potentials and D-wave recordings in the recent years). The patients were positioned depending on the location of the tumor: A semi-sitting position was performed for tumors of the upper and middle cervical region and prone position for lesions of the lower cervical and thoracic region including the medullary conus. The extent of tumor resection was judged upon the operative report.
IDSCT were classified into two groups: Intramedullary SCT (IMSCT) and extramedullary SCT (EMSCT). There were 29 (58%) patients suffering from EMSCT and 21 (42%) from IMSCT. The functional outcome was correlated with the histological features, tumor location, and the extension of the tumor. Surgical mortality was referred to death from any cause within 30 days of surgery, whereas the IDSCT mortality was referred to the cases issuing from IDSCT progression.
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics 22 (IBM Corporation, USA). Interval-scaled data were expressed as mean and standard deviations and nominal data were expressed as absolute numbers and valid percent.
Correlation analyses were conducted using Spearman's Rho due to not normal distribution of the data. P value of ≤0.05 was considered as statistically significant.
The investigated cohort consisted predominantly of males (n = 32, 64%), the mean age was 10.3 years (range 6 months–19 years). There were no differences in age and sex between patients with IMSCT and EMSCT.
Clinical presentation
The mean duration between the first symptoms and diagnosis was 2.8 months (from incidental finding up to 1 year) in patients with IMSCT and 8.1 ± 9.9 months (from incidental finding up to 3 years) in cases with EMSCT. The initial symptoms at the time of admission were also different between both groups. The most common symptom in patients who suffered from EMSCT was back or neck pain. Monoparesis was more often in patients with IMSCT (62%), compared to only nine cases in the EMSCT-group (31%). The rates of gait disturbance were comparable in both groups with 19% and 21% for EMSCT and IMSCT, respectively. A detailed description of symptoms is shown in Table 1 .
Table 1
Symptoms at admission
More often IMSCT were located in the cervical part of the spine, whereas EMSCT were more typical for lumbar spine. Tumor locations are presented in Figure 1 .
Figure 1
Level of lesion depended on tumor location
Histopathology
Histological findings of IMSCT and EMSCT are shown in Table 2 . The most common primary IMSCT were astrocytomas (12, 57%) followed by metastases arising from intracranial medulloblastomas (7 patients, 29%). In turn, neurinomas (10, 34%) and lipomas (9, 31%) represented the most frequent lesions of the intradural extramedullary compartment. Most tumors were benign (Grade I–II, 78%) and only 11 patients (22%) had high-grade tumors according to the World Health Organization (WHO)-classification.
Table 2
Histopathology and medullary location of IDSCTs
Neurosurgical treatment
Forty-four patients (88%) underwent surgical treatment of IDSCT. In five cases with intramedullary metastases, intrathecal chemotherapy was administered through an Ommaya reservoir. One patient with intradural extramedullary lipoma underwent conservative management.
Laminoplasty was the standard surgical approach for intraspinal and intradural lesions (27 patients, 61%). Laminectomy was used in the early period of this study for intradural extramedullary tumors. Stabilization of the spine was necessary in only one case of a 14-year-old boy suffering from a spinal metastasis of a colon cancer.
Complete surgical removal was 71% for IMSCT and 59% for EMSCT, respectively.
The surgical morbidity rate was 2%. A postoperative wound infection was observed in one case, and a temporary percutaneous tracheotomy was necessary in 2 patients. Two patients developed pneumonia, which were treated successfully with antibiotic medication.
Illustrative case
A 14-year-old girl suffered from bilateral arm weakness for 1 year. Gait disturbances or other neurological deficits were not present. An MRI was performed revealing an IMSCT from C5 to C7 with cystic and solid tumor compartments without contrast enhancement. The tumor was removed via a laminotomy of the vertebral arch from C5 to C7 under intraoperative electrophysiological monitoring. The tumor was removed incompletely due to the lack of a clear plane of cleavage. Histopathological examination revealed a fibrillary astrocytoma according to the WHO Grade II. Sensory deficits in arms and legs were observed postoperatively, but improved until discharge and during the postoperative rehabilitation continuously [ Figure 2 ].
Figure 2
A 14-year-old girl with an intramedullary cystic fibrilar astrocytoma World Health Organization II from C5 to C7. (a and b) Preoperative T2 and T1 with contrast enhancement in sagittal view of the tumor and interestingly the vertebral body deformation from C3 to C6 due to the slow tumor growing (arrow), (c-e) T2 in axial view preoperatively, (f) 5 months after surgery with tumor rests in the apical and caudal part of the initial tumors, (g) T2 sagittal view 10 months after surgery and (h) 6 years after surgery, only the postoperative defect of the initial tumor were seen
Early postoperative outcome
In summary, 17 patients (38%) developed transitory neurological symptoms (new sensory deficits, paresis, or gait disturbances) directly after the surgery. However, these symptoms were mostly reversible, so that the postoperative Frankel score at discharge was comparable with the preoperative score.
Postoperative transitory neurological deterioration was predominantly observed in IMSCT patients (n = 12). Among them, only 1 patient with an IMSCT suffered from a persistent postoperative neurological deterioration from Fankel Grade E to C. In turn, EMSCT patients were more likely to improve in their neurological condition postoperatively [ Table 3 ].
Table 3
Functional outcome according to the Frankel classification grading system at different time points
Follow-up
The mean duration of follow-up was 3.5 years (range 3 months–10 years). Spinal deformity after surgery was not detected except for 1 female patient after laminectomy. This patient had to be re-operated for spine stabilization using dorsal transpedicular screw and rod fixation. In other case with the removal of a spinal lipoma at the age of 1 year and 11 years later repeated surgery with detethering was required because of the development of a tethered cord.
Short-term follow-up at 12 months after discharge was available for 33 (66%) patients (18 with EMSCT, 15 with IMSCT). Nine (50%) patients with EMSCT recovered in this time up to the preoperative Frankel score. Among IMSCT patients, the complete recovery could be achieved in only 3 patients (20%).
At the 12 months follow-up, the Frankel score of the EMSCT-group was higher than the Frankel score of the IMSCT-group [ Table 3 ].
The WHO grading of the tumor was significantly correlated with the functional outcome at discharge (P = 0.017) and at later follow-ups (P = 0.02). Moreover, there was a statistically significant positive correlation between the preoperative Frankel score and the last follow-up (P = 0.002) [ Figure 3 ].
Figure 3
Frankel score in follow-up Exitus letalis in the extramedullay group: 2, intramedullary group: 4
Recurrence
Tumor recurrence was seen in 7 (14%) patients during the whole observational time. In all of these cases, the initial tumor resection was performed subtotally. Four of them (57%) had IMSCT. The detailed description of the cases with recurrent IDSCT is given in Table 4 .
Table 4
Recurrences
Mortality
Five individuals (10%) died during the follow-up due to the tumor progress. These cases include two 14-year-old patients with intramedullary anaplastic astrocytoma (WHO Grade III) who died 2 and 5 years after the surgery, respectively. Two other patients died 1 and 3 years after surgery, suffering from metastasis from colon carcinoma and medulloblastoma. Finally, a 14-year-old male suffered from malignant peripheral nerve sheath tumor and died 2 months postoperatively.
After the implementation of a modern neuroradiological imaging and microsurgical techniques, better functional outcome after IDSCT surgery could be achieved, especially for IMSCT.[6]
The specific aspects of perioperative care for spinal tumors and spine trauma in the mature spine have been widely described in the modern literature.[361117] In the present study, we addressed the challenges and clinical impact of modern microsurgery in the pediatric population with IDSCT.
Clinical presentation
The symptoms of IDSCT in children and young adults are attributed to spinal cord compression, and include pain and motor weakness of the extremities. The most common symptoms of patients with EMSCT were pain/back pain (41%). Patients suffering from IMSCT presented more common with the motor weakness of extremities (52%). Constantini et al.[ 7 ] have also reported the motor regression as the leading symptom in 65.2% of intramedullary tumors in children and young adults.
In our cohort, the mean duration of the symptoms before admission was 8.1 months for EMSCT and 2.8 months for IMSCT. Observation is partially based on spinal cord plasticity allowing considerable tumor progression without the development of meaningful neurological deficits.[ 11 ] Furthermore, the diagnostic delay which is common in pediatric malignancies,[ 33 ] could also have contributed to the longer duration of clinical symptoms in the preoperative stage in our cohort.
Such long clinical “tolerability” of IDSCT explain the fact that Dincer et al.[ 11 ] reported about 4.8% scoliosis as an initial symptom and emphasized that it could be related to delayed diagnosis. However, the deformities of the spine or scoliosis are not seen in the patients with IDSCT in our series. We agree with the most of authors[ 6 7 13 16 26 ] who concluded that the residual disability after the treatment is rather attributed to the delay in the diagnosis, than due to the surgical technique or postoperative tumor progression.[ 11 35 36 ]
Surgery
Due to the high-risk of secondary spinal deformity in the pediatric patients,[ 10 28 41 42 ] the surgical approach remains a matter of debate in the treatment of IDSCT in children and young adults. The use of laminoplasty has not been proven to be superior with regard to the development of spinal deformity after surgery compared to laminectomy.[ 1 28 38 41 42 ] However, it seems to have a positive effect on wound healing and the reduction of cerebrospinal fluid leakages.[ 38 ] Furthermore, the pathogenesis of spinal deformity after IMSCT surgery is not only the result of the selected surgical approach, but rather an effect from the underlying neuromuscular dysfunction.[ 2 ]
On the other hand, the use of prophylactic fusion has certain challenges in the pediatric population, such as the risk of neuromuscular scoliosis and “crank-shaft”- deformity due to the continued growth of the anterior spine column, a high rate of pseudarthrosis and the difficulties of screw placement based on the small pedicles.[ 27 29 ]
In our opinion, the reposition of the posterior arches with micro plates may prevent the development of postoperative scare and, therefore, protects the nervous structures, but does not prevent the development of spinal deformities. Our current surgical strategy consists of the use of laminoplasty without prophylactic fusion and continuous clinical and radiological follow-up that may warrant the timely detection of the secondary spinal deformity in younger individuals.
The extension of the tumor resection depends on tumor histology and location of the tumor.[ 14 17 ] Although several authors reported a series with complete tumor removal and good postoperative functional outcome,[ 7 14 16 37 ] others pointed out the limitations of the surgery caused by the absence of a clear plane of cleavage, resulting in partial resection, decompression, or biopsy only for diagnosis.[ 26 ]
In our cohort, the majority of EMSCT were resected completely, except of lipomas and intradural extramedullary metastasis. Intradural meningiomas were completely resected, whereas in one case a Grade I meningioma was recurrent and, therefore, re-surgery was performed.
Astrocytomas are characterized as infiltrative growing tumors, without clear border to the spinal cord tissue and, therefore, often leading to an incomplete tumor removal.[ 4 18 ] Whereas ependymomas are tumors which are recognized as resectable lesions due to a clearly defined plane of dissection.[ 39 ] Safaee et al.[ 39 ] noted that the extent of resection of spinal ependymoma is an important prognostic factor. In our cohort, the complete removal of IMSCT has been achieved in 60% of the cases, whereat ependymomas were completely resected in 90%. Constantini et al.[ 7 ] reported in his large series about 164 patients with IMSCT (age between 6 months and 21 years). They observed that radical surgery for low-grade IMSCTs could be performed with an acceptable risk and moderate functional outcome. These findings correlated with our series. Furthermore, the postoperative functional performance is mainly determined by the preoperative deficits.[ 7 ] Therefore, IMSCTs are potentially excisable lesions, both at presentation and if they recur.[ 2 6 7 8 9 11 14 16 23 26 30 31 ] The treatment management of primary malignant spinal cord astrocytomas is not clearly determined in the current literature, neither for children nor in adults.[ 6 7 40 ]
Functional outcome
Functional outcome after surgery has correlated strongly with the preoperative neurological status, both in EMSCT and in IMSCT patients. Postoperative temporary worsening was seen more likely in patients with IMSCT. We agree with the hypothesis of Epstein,[ 14 ] that particularly the patients with large, long-standing IMSCT may be attributed to thinning of the spinal parenchyma.
Interestingly, at the time of admission the functional grade was good in 75% of EMSCT and 81% of IMSCT patients. Both groups resulted in an improvement or total functional recovery at discharge.
There was no surgery-related mortality in our study. The overall mortality from tumor progression was 10% (n = 8), mainly due to leptomeningeal metastases. Previous reports based upon intramedullary tumors described even higher rates of mortality due to tumor progression (up to 22%).[ 5 7 ] In addition, the overall recurrence rate of 14% in our study is also considerably lower, as compared to earlier series with recurrent intramedullary tumors in 35% of the patients.[ 11 ]
In summary, the postoperative functional outcome in the presented cohort is comparable with the previous reports.[ 3 4 14 18 25 ] The use of modern neurosurgical techniques, as well as advanced perioperative and neuro-oncological management may contribute to further improvement of the functional outcome of younger individuals with IDSCT.
Study limitations
This study presents a retrospective, single-institution analysis and, therefore, faces certain limitations. Retrospective analysis introduces recall bias and difficulty for controlling of confounders. However, randomized prospective studies are difficult for surgical diseases, particularly for pediatric IDSCTs. Then, the histopathological and age heterogeneity of the presented data should also be mentioned as study limitations. Finally, due to the nonuniformly performed postoperative clinical and radiological follow-up, our data do not allow any assumption about the correlations between the surgical radicality and postoperative outcome.
IDSCTs are good candidates for radical surgical treatment. As to the surgical technique, we advocate laminotomy to reduce the secondary surgery-related complications. Multicenter prospective database may be helpful in the development of optimal surgical strategies in children and young adults with IDSCTs.
1. Ahmed R, Menezes AH, Awe OO, Mahaney KB, Torner JC, Weinstein SL. Long-term incidence and risk factors for development of spinal deformity following resection of pediatric intramedullary spinal cord tumors. J Neurosurg Pediatr. 2014. 13: 613-21
2. Ahmed R, Menezes AH, Awe OO, Torner JC. Long-term disease and neurological outcomes in patients with pediatric intramedullary spinal cord tumors. J Neurosurg Pediatr. 2014. 13: 600-12
3. Arseni C, Horvath L, Iliescu D. Intraspinal tumours in children. Psychiatr Neurol Neurochir. 1967. 70: 123-33
4. Babu R, Hazzard MA, Huang KT, Ugiliweneza B, Patil CG, Boakye M. Outcomes of percutaneous and paddle lead implantation for spinal cord stimulation: A comparative analysis of complications, reoperation rates, and health-care costs. Neuromodulation. 2013. 16: 418-26
5. Baysefer A, Akay KM, Izci Y, Kayali H, Timurkaynak E. The clinical and surgical aspects of spinal tumors in children. Pediatr Neurol. 2004. 31: 261-6
6. Constantini S, Houten J, Miller DC, Freed D, Ozek MM, Rorke LB. Intramedullary spinal cord tumors in children under the age of 3 years. J Neurosurg. 1996. 85: 1036-43
7. Constantini S, Miller DC, Allen JC, Rorke LB, Freed D, Epstein FJ. Radical excision of intramedullary spinal cord tumors: Surgical morbidity and long-term follow-up evaluation in 164 children and young adults. J Neurosurg. 2000. 93: S183-93
8. Cooper PR, Epstein F. Radical resection of intramedullary spinal cord tumors in adults. Recent experience in 29 patients. J Neurosurg. 1985. 63: 492-9
9. Cristante L, Herrmann HD. Surgical management of intramedullary spinal cord tumors: Functional outcome and sources of morbidity. Neurosurgery. 1994. 35: 69-74
10. de Jonge T, Slullitel H, Dubousset J, Miladi L, Wicart P, Illés T. Late-onset spinal deformities in children treated by laminectomy and radiation therapy for malignant tumours. Eur Spine J. 2005. 14: 765-71
11. Dincer F, Dincer C, Baskaya MK. Results of the combined treatment of paediatric intraspinal tumours. Paraplegia. 1992. 30: 718-28
12. Ekelund L, Cronqvist S. Roentgenological changes in spinal malformations and spinal tumours in children. Radiologe. 1973. 13: 541-6
13. el-Mahdy W, Kane PJ, Powell MP, Crockard HA. Spinal intradural tumours: Part I – Extramedullary. Br J Neurosurg. 1999. 13: 550-7
19. Frankel HL, Hancock DO, Hyslop G, Melzak J, Michaelis LS, Ungar GH. The value of postural reduction in the initial management of closed injuries of the spine with paraplegia and tetraplegia. I. Paraplegia. 1969. 7: 179-92
20. Friedman WA, Grundy BL. Monitoring of sensory evoked potentials is highly reliable and helpful in the operating room. J Clin Monit. 1987. 3: 38-44
21. Gowers WR, Horsley V. A case of tumour of the spinal cord. Removal; recovery. Med Chir Trans. 1888. 71: 377-430
22. Gunzburg R, Szpalski M, Aebi M.editors. Vertebral Tumors. Philadelphia, PA, USA: Lippincott William and Wilkins; 2008. p.
23. Huisman TA. Pediatric tumors of the spine. Cancer Imaging. 2009. 9: S45-8
25. Jenkinson MD, Simpson C, Nicholas RS, Miles J, Findlay GF, Pigott TJ. Outcome predictors and complications in the management of intradural spinal tumours. Eur Spine J. 2006. 15: 203-10
26. Kane PJ, el-Mahdy W, Singh A, Powell MP, Crockard HA. Spinal intradural tumours: Part II – Intramedullary. Br J Neurosurg. 1999. 13: 558-63
27. Katsumi Y, Honma T, Nakamura T. Analysis of cervical instability resulting from laminectomies for removal of spinal cord tumor. Spine (Phila Pa 1976). 1989. 14: 1171-6
28. Klekamp J. Treatment of intramedullary tumors: Analysis of surgical morbidity and long-term results. J Neurosurg Spine. 2013. 19: 12-26
29. Lapinksy AS, Richards BS. Preventing the crankshaft phenomenon by combining anterior fusion with posterior instrumentation. Does it work?. Spine (Phila Pa 1976). 1995. 20: 1392-8
30. Li TY, Chu JS, Xu YL, Yang J, Wang J, Huang YH. Surgical strategies and outcomes of spinal ependymomas of different lengths: Analysis of 210 patients: Clinical article. J Neurosurg Spine. 2014. 21: 249-59
31. Lin Y, Jea A, Mekonian SC, Lan S. Treatment of pediatric grad II spinal ependymonas: A population-based study. J Neurosurg Petiatr. 2014. 15: 243-9
32. Rubinstein LJ.editors. Tumors of neuroglial cells. Washington, DC: Armed Forces Institute of Pathology; 1972. p.
33. Loh AH, Aung L, Ha C, Tan AM, Quah TC, Chui CH. Diagnostic delay in pediatric solid tumors: A population based study on determinants and impact on outcomes. Pediatr Blood Cancer. 2012. 58: 561-5
34. Long RR, Wirth FP. Reversible somatosensory evoked potential changes with neodymium: Yttrium-aluminum-garnet laser use. Neurosurgery. 1987. 21: 465-7
35. Manzano G, Green BA, Vanni S, Levi AD. Contemporary management of adult intramedullary spinal tumors-pathology and neurological outcomes related to surgical resection. Spinal Cord. 2008. 46: 540-6
36. Matson DD, Tachdjian MO. Intraspinal tumors in infants and children; review of 115 cases. Postgrad Med. 1963. 34: 279-85
37. McCormick PC, Torres R, Post KD, Stein BM. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990. 72: 523-32
38. McGirt MJ, Garcés-Ambrossi GL, Parker SL, Sciubba DM, Bydon A, Wolinksy JP. Short-term progressive spinal deformity following laminoplasty versus laminectomy for resection of intradural spinal tumors: Analysis of 238 patients. Neurosurgery. 2010. 66: 1005-12
39. Safaee M, Oh MC, Mummaneni PV, Weinstein PR, Ames CP, Chou D. Surgical outcomes in spinal cord ependymomas and the importance of extent of resection in children and young adults. J Neurosurg Pediatr. 2014. 13: 393-9
40. Wong AP, Dahdaleh NS, Fessler RG, Melkonian SC, Lin Y, Smith ZA. Risk factors and long-term survival in adult patients with primary malignant spinal cord astrocytomas. J Neurooncol. 2013. 115: 493-503
41. Yasuoka S, Peterson HA, Laws ER Jr, MacCarty CS. Pathogenesis and prophylaxis of postlaminectomy deformity of the spine after multiple level laminectomy: Difference between children and adults. Neurosurgery. 1981. 9: 145-52
42. Yasuoka S, Peterson HA, MacCarty CS. Incidence of spinal column deformity after multilevel laminectomy in children and adults. J Neurosurg. 1982. 57: 441-5
43. Zieger M, Dörr U. Pediatric spinal sonography. Part I: Anatomy and examination technique. Pediatr Radiol. 1988. 18: 9-13
44. Zieger M, Dörr U, Schulz RD. Pediatric spinal sonography. Part II: Malformations and mass lesions. Pediatr Radiol. 1988. 18: 105-11
How to cite this article: Gadgil N, Hansen D, Barry J, Chang R, Lam S. Posterior fossa syndrome in children following tumor resection: Knowledge update. Surg Neurol Int 11-Mar-2016;7:
A 2-year-old female presented with a 2-month history of recalcitrant vomiting followed by ataxia and lethargy. Computed tomography (CT) scan of the head showed a large solid and cystic mass in the cerebellar vermis with severe hydrocephalus. Magnetic resonance imaging (MRI) demonstrated a 6.0 cm × 4.4 cm × 4.2 cm uniformly enhancing mass concerning for medulloblastoma with no evidence of spinal metastasis. She underwent external ventricular drain (EVD) placement and uneventful posterior fossa craniotomy with gross total resection of the lesion [ Figure 1 ]. For surgical exposure, the lower two-thirds of the vermis was split in the midline. Pathology confirmed classic medulloblastoma M0. Postoperatively, the patient displayed mutism, left-sided dysmetria, truncal/gait ataxia, and mild generalized hypotonia. The patient had postoperative hydrocephalus and required ventriculoperitoneal (VP) shunt placement. Her mutism symptoms improved significantly by 1 month, with continuing speech therapy and occupational therapy. At her 2-month follow-up, she demonstrated persistent mild dysarthria, ataxia, and left-sided dysmetria. The patient completed high-dose chemotherapy and radiation therapy. At last follow-up, 45 months after tumor resection, she had entered age-appropriate first grade academics, but continues to have mild dysarthria, dysmetria, hypotonia, and wide-based gait.
Figure 1
(a) Axial and sagittal T1 postcontrast magnetic resonance images. Large heterogeneous tumor, dorsal to the brainstem and occupying much of the posterior fossa with resulting obstructive hydrocephalus. (b) Postresection images in similar planes showing gross total resection of tumor and resulting decompression of brain stem and ventricular system
Case 2
A 2-year-old boy presented with a 3-week history of progressive headache and daily vomiting. A CT scan revealed a solid mass in the fourth ventricle with moderate obstructive hydrocephalus. Presurgical MRI confirmed a 6.1 cm × 4.9 cm × 4.3 cm mass centered in the fourth ventricle and extending out the foramen of Luschka, consistent with an ependymoma. EVD placement and resection of the tumor were carried out in the same setting [ Figure 2 ]. Posterior fossa craniotomy was performed and gross total resection was achieved; however, the tumor presented itself posteriorly and no splitting of the vermis was required. Cranial nerve monitoring was utilized due to the intimate nature of the tumor to the brainstem. Pathology was consistent with ependymoma. Postoperatively, the patient had complete mutism, but he otherwise demonstrated good neurologic function. Due to continued hydrocephalus, a VP shunt was placed several days after the original operation. By 2 months, the patient was speaking a few words and by 6 months had returned to his age-appropriate neurologic baseline. He did receive proton beam radiation therapy to the tumor bed.
Figure 2
(a) Fluid-attenuated inversion recovery axial and T2 coronal magnetic resonance images. Large nonenhancing tumor, wrapping ventral to the brainstem and occupying the posterior fossa with resultant hydrocephalus. (b) Same sequence postresection images showing gross total resection of tumor, with mild reduction in ventricular caliber
Cerebellar mutism syndrome (CMS) refers to constellation of symptoms noted most commonly following surgery for posterior fossa tumors in the pediatric population. Mutism is a prominent, though not exclusive, characteristic of the syndrome and was first described in 1985 by Rekate et al.[27] In their paper, the authors described six children ranging from 1 to 6 years of age who developed complete absence of speech following resection of posterior fossa masses. Loss of speech was transient in all patients in this series, though many subsequent descriptions has been long lasting.[153132] The term “syndrome” refers to a set of signs and symptoms observed to have correlation, but without clear understanding of underlying common pathogenesis. Although hundreds of articles now have been published describing what we commonly refer to as “posterior fossa syndrome,” its unclear pathophysiology and pervasive consequences warrant further investigation.
CMS occurs in 8–24% of children following resection of posterior fossa masses.[242935] Other uncommon though interesting pathophysiologic causes of this syndrome include trauma, strokes, and infection.[491423] Rarely, CMS has been described in adults.[734] The syndrome is characterized most prominently by the absence or reduction in speech within 1–2 days of surgery without alteration in the level of consciousness.[61228] Patients may retain the ability to produce speech immediately following surgery and become mute within an average of 2 days, but up to 7 days postoperatively.[1229] Mutism is frequently accompanied by profound axial hypotonia and ataxia. One of the hallmarks of CMS is that long tract signs are not present; although hemiparesis has been described in the setting of CMS, the gradual return of motor function is typically accompanied by severe ataxia, suggesting that the paresis was, in fact, a result of cerebellar dysfunction.[15] Signs of brainstem dysfunction may be present, most commonly dysarthria,[28] dysphagia,[20] and abducens or facial nerve palsies. Mood lability or so-called “pseudobulbar affect,”[122028] cognitive deficits,[2837] and urinary incontinence or retention[33] may also be present. Poor oral intake and apathy are common. The syndrome has a broad spectrum of severity, ranging from mild to completely disabling symptoms.
The duration of mutism varies widely, with an average of approximately 8 weeks, but a range of 4 days to 5 months.[15] Although many consider CMS temporary due to the transience of mutism in most patients, the majority of patients experience persistent symptoms that may be debilitating. The pattern of speech return has been described in one long-term study as follows: Initially, the child speaks in single word and then progresses to full sentences. Speech is initially slow, quiet, and monotonous, with or without dysarthria. Long-term persistent dysarthria is common, and speech often remains slow and lacking in spontaneity.[293239] This cerebellar ataxic dysarthria is primarily a motor impairment. Meta-analysis showed that even after the initial mutism had resolved, 68% of patients had residual motor speech deficits a year after surgery.[15] Emotional lability is typically transient.[19] Other long-term sequelae include apraxia, ataxia, linguistic, memory, or behavioral disabilities.[153132] Neuropsychological tests for children with CMS, compared to a control group, found statistically significant deficits in intellect, processing speed, attention, working memory, auditory processing, and spatial relations 12 months after surgery.[22] Functional prognosis correlates with the initial severity of symptoms as well as the duration of symptoms after surgery. Patients with mutism for greater than 4 weeks are at an increased risk of speech and language pathologies at 1-year follow-up.[1729] In some cases, patients make rapid full recovery.[32]
Despite collective efforts to describe the pathophysiological mechanism of CMS, the answer remains elusive. Several predisposing risk factors have been observed. Tumor pathology has proven the most predictive, with medulloblastoma patients experiencing a two- to three-fold increased chance of developing CMS as compared to other posterior fossa tumors.[51539] Midline tumors, particularly those involving the vermis and those high in the fourth ventricle, present higher risk;[535] tumor involvement in or compression of the brainstem also carries a higher risk of postoperative CMS.[81829] Some studies suggest that a vermian incision may pose a greater risk of CMS,[510] though this is not universally reported. Studies have shown conflicting results regarding the correlation of tumor size to the development of CMS.[5152938] Patients presenting with hydrocephalus in some series have a higher incidence of CMS,[36] though this has not been borne out in every study.[33] Patients with preoperative language impairment also have dramatically higher rates of CMS.[33] However, CMS occurs in only a subset of patients even with high preoperative similarity.
Although there is no clear consensus in the literature, many associate damage to the dentato-thalamo-cortical pathway with CMS.[1219252836] The cerebral cortex delivers input to the cerebellum along the cortico-ponto-cerebellar pathway that eventually synapses on Purkinje cells of the cerebellum. These cells synapse on the deep cerebellar nuclei, from which efferent signals travel throughout the central nervous system. The dentate nuclei act as integration centers between the cerebellum and cortex, and they send efferent signals back to the cerebral cortex via dentate-thalamo-cortical pathway travelling through the superior cerebellar peduncle. This pathway plays a role in planned motor activity, coordination, and movement; it is also thought to modulate cognition and behavior. It is proposed that damage to efferent white matter pathways travelling through the superior cerebellar peduncle is responsible for CMS, particularly in cases of bilateral damage.[2125] Damage to these pathways secondarily creates a phenomenon called diaschisis, in which cerebral cortical areas that receive input from cerebellar pathways become hypofunctional once input pathways are compromised. Two studies have shown that release of tumor tension from the superior cerebellar peduncle and subsequent inability to perceive the superior cerebellar peduncle white matter tracts on diffusion tensor imaging were predictive for the development of CMS.[1821]
Damage to the vermis may also be important in the development of CMS. The vermis is implicated in speech initiation; while splitting of the inferior third of the vermis is not thought to increase CMS, damage to the superior vermis is considered to be a higher risk.[33] The vermis is posited to function like the limbic system of the cerebellum and is involved in complex social behavior mechanisms, emotions, and ability to plan. Puget et al. reported that the radiographic degree of damage to the dentate nuclei and the inferior vermis as seen on MRI directly correlates with the severity of cerebellar deficits.[26]
Bilateral dentate nuclei damage is also theorized to result in CMS, a theory backed by early work demonstrating mutism following stereotactic lysis of the dentate nuclei for dyskinesia.[13] The preponderance of midline tumors in CMS patients suggests involvement of the dentate nuclei in the development of this syndrome.
Direct damage to cerebellar neuronal pathways fails to explain why many patients are initially intact postoperatively and develop deficits after a few days, a finding that has led to speculation about other mechanisms. The onset of mutism coincides with the peak timing of postoperative edema;[25] some have reported that postoperative edema involving the dentate nuclei and progressive swelling of paramedian structures explains the delayed onset of mutism. However, edema alone fails to explain why deficits are frequently permanent.[22] Some authors have theorized that deficits in cerebellar perfusion caused by vascular manipulation or vasospasm may also account for the delay in the onset of symptoms.[2] While one study has found improvement of perfusion on single photon emission CT, correlated with improvement in symptoms,[12] others have directly refuted this theory.[8]
To date, no specific treatment has been found for CMS other than supportive care. At our institution, many patients with this syndrome have been referred for intensive inpatient rehabilitation. Patients may require gastrostomy tube placement and intensive speech, physical, and occupational therapy. Although our outcomes have been positive, recovery is gradual and may remain incomplete. For patients with primarily dysarthric speech disorders, exercises focused on coordination of sensorimotor integration should be emphasized. Other patients may have an apraxic language disorder, in which procedural memory and recognition of sensory stimuli is defective; this manifests in slow, monotone speech. Emphasis for these patients should be placed on the awareness of visual and auditory stimuli and planning of sound sequences.[33]
Several groups have reported single patient trials of pharmaceutical therapies for CMS, including steroids, fluoxetine, bromocriptine, or zolpidem.[131130] While each had reported a positive result, the gradual improvement of the syndrome with time makes it difficult to attribute therapeutic effect to these medications.
Our lack of understanding of the precise pathophysiologic mechanism of CMS makes it difficult to accurately forecast which patients will develop the syndrome. While several predictive factors have been found, none can be used with certainty. Therefore, our hope at this time is prevention of damage from aggressive tumor resection.[15] In the face of malignant pediatric tumors, radical resections often confer superior prognosis. Some have proposed that a surgical approach that spares dissection of the vermis, specifically the telovelar approach, may reduce the risk of CMS. While one study using this method had no occurrences of CMS in 16 patients,[10] another found a postoperative incidence of CMS with this approach of 30% in a series of 20 patients.[40] CMS has been reported after both unilateral and bilateral telovelar dissections.[33] The relative importance of splitting the vermis versus damage to deep midline cerebellar structures from surgical manipulation is unclear. Hermann et al. proposed a transventricular supracerebellar approach to the fourth ventricle; in their series, there was no occurrence of CMS.[16] However, this approach may only be effective for tumors high in the fourth ventricle. Ultrasound-assisted surgery may assist in the safety of resections by decreasing the amount of retraction on cerebellar structures during exploration.[33] In summary, there are conflicting theories regarding the development of CMS; agreement across various studies is to avoid retraction and manipulation, particularly on deep midline cerebellar structures.
CMS is a common but devastating complication of posterior fossa surgery in children. While the mutism itself is often transient, permanent sequelae are common. The precise pathophysiology of this disease remains unknown, and treatment focuses on supportive care symptoms. At Texas Children's Hospital, multidisciplinary evaluation and treatment are integral to brain tumor care. Physical medicine and rehabilitation, neurology, ophthalmology, and neuro-oncology teams evaluate and follow patients; those with continuing therapy needs transition to intensive inpatient rehabilitation after surgery. Further investigation as to the underlying mechanism of CMS likely holds the promise of prevention and treatment of this syndrome.
1. Adachi J, Nishikawa R, Hirose T, Matsutani M. Mixed neuronal-glial tumor of the fourth ventricle and successful treatment of postoperative mutism with bromocriptine: Case report. Surg Neurol. 2005. 63: 375-9
2. Aguiar PH, Plese JP, Ciquini O, Marino R. Transient mutism following a posterior fossa approach to cerebellar tumors in children: A critical review of the literature. Childs Nerv Syst. 1995. 11: 306-10
3. Akhaddar A, Salami M, El Asri AC, Boucetta M. Treatment of postoperative cerebellar mutism with fluoxetine. Childs Nerv Syst. 2012. 28: 507-8
4. Baillieux H, Weyns F, Paquier P, De Deyn PP, Mariën P. Posterior fossa syndrome after a vermian stroke: A new case and review of the literature. Pediatr Neurosurg. 2007. 43: 386-95
5. Catsman-Berrevoets CE, Van Dongen HR, Mulder PG, Paz y Geuze D, Paquier PF, Lequin MH. Tumour type and size are high risk factors for the syndrome of “cerebellar” mutism and subsequent dysarthria. J Neurol Neurosurg Psychiatry. 1999. 67: 755-7
6. Clerico A, Sordi A, Ragni G, Festa A, Cappelli C, Maini CL. Brief report: Transient mutism following posterior fossa surgery studied by single photon emission computed tomography (SPECT). Med Pediatr Oncol. 2002. 38: 445-8
7. De Smet HJ, Mariën P. Posterior fossa syndrome in an adult patient following surgical evacuation of an intracerebellar haematoma. Cerebellum. 2012. 11: 587-92
8. Doxey D, Bruce D, Sklar F, Swift D, Shapiro K. Posterior fossa syndrome: Identifiable risk factors and irreversible complications. Pediatr Neurosurg. 1999. 31: 131-6
9. Drost G, Verrips A, Thijssen HO, Gabreëls FJM. Cerebellar involvement as a rare complication of pneumococcal meningitis. Neuropediatrics. 2000. 31: 97-9
10. El-Bahy K. Telovelar approach to the fourth ventricle: Operative findings and results in 16 cases. Acta Neurochir (Wien). 2005. 147: 137-42
11. El-Nabbout B, DeLong G.editors. Treatment of cerebellar mutism with fluoxetine: Report on two patients. Annals of Neurology. New York: Wiley-Liss, Div John Wiley & Sons Inc; 2002. p.
12. Ersahin Y, Mutluer S, Cagli S, Duman Y. Cerebellar mutism: Report of seven cases and review of the literature. Neurosurgery. 1996. 38: 60-5
13. Fraioli B, Guidetti B. Effects of stereotactic lesions of the dentate nucleus of the cerebellum in man. Appl Neurophysiol. 1975. 38: 81-90
14. Fujisawa H, Yonaha H, Okumoto K, Uehara H, Ie T, Nagata Y. Mutism after evacuation of acute subdural hematoma of the posterior fossa. Childs Nerv Syst. 2005. 21: 234-6
15. Gelabert-González M, Fernández-Villa J. Mutism after posterior fossa surgery. Review of the literature. Clin Neurol Neurosurg. 2001. 103: 111-4
16. Hermann EJ, Rittierodt M, Krauss JK. Combined transventricular and supracerebellar infratentorial approach preserving the vermis in giant pediatric posterior fossa midline tumors. Neurosurgery. 2008. 63: ONS30-5
17. Levisohn L, Cronin-Golomb A, Schmahmann JD. Neuropsychological consequences of cerebellar tumour resection in children: Cerebellar cognitive affective syndrome in a paediatric population. Brain. 2000. 123: 1041-50
18. McMillan HJ, Keene DL, Matzinger MA, Vassilyadi M, Nzau M, Ventureyra EC. Brainstem compression: A predictor of postoperative cerebellar mutism. Childs Nerv Syst. 2009. 25: 677-81
19. Morris EB, Phillips NS, Laningham FH, Patay Z, Gajjar A, Wallace D. Proximal dentatothalamocortical tract involvement in posterior fossa syndrome. Brain. 2009. 132: 3087-95
20. Mortimer DS. Clinical case study: A 4-year-old boy with posterior fossa syndrome after resection of a medulloblastoma. J Neurosci Nurs. 2011. 43: 225-9
21. Ojemann JG, Partridge SC, Poliakov AV, Niazi TN, Shaw DW, Ishak GE. Diffusion tensor imaging of the superior cerebellar peduncle identifies patients with posterior fossa syndrome. Childs Nerv Syst. 2013. 29: 2071-7
22. Palmer SL, Hassall T, Evankovich K, Mabbott DJ, Bonner M, Deluca C. Neurocognitive outcome 12 months following cerebellar mutism syndrome in pediatric patients with medulloblastoma. Neuro Oncol. 2010. 12: 1311-7
23. Papavasiliou AS, Kotsalis C, Trakadas S. Transient cerebellar mutism in the course of acute cerebellitis. Pediatr Neurol. 2004. 30: 71-4
25. Pollack IF, Polinko P, Albright AL, Towbin R, Fitz C. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: Incidence and pathophysiology. Neurosurgery. 1995. 37: 885-93
26. Puget S, Boddaert N, Viguier D, Kieffer V, Bulteau C, Garnett M. Injuries to inferior vermis and dentate nuclei predict poor neurological and neuropsychological outcome in children with malignant posterior fossa tumors. Cancer. 2009. 115: 1338-47
28. Riva D, Giorgi C. The cerebellum contributes to higher functions during development: Evidence from a series of children surgically treated for posterior fossa tumours. Brain. 2000. 123: 1051-61
29. Robertson PL, Muraszko KM, Holmes EJ, Sposto R, Packer RJ, Gajjar A. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: A prospective study by the Children's Oncology Group. J Neurosurg. 2006. 105: 444-51
30. Shyu C, Burke K, Souweidane MM, Dunkel IJ, Gilheeney SW, Gershon T. Novel use of zolpidem in cerebellar mutism syndrome. J Pediatr Hematol Oncol. 2011. 33: 148-9
32. Steinbok P, Cochrane DD, Perrin R, Price A. Mutism after posterior fossa tumour resection in children: Incomplete recovery on long-term follow-up. Pediatr Neurosurg. 2003. 39: 179-83
33. Tamburrini G, Frassanito P, Chieffo D, Massimi L, Caldarelli M, Di Rocco C. Cerebellar mutism. Childs Nerv Syst. 2015. 31: 1841-51
34. van Baarsen K, Kleinnijenhuis M, Konert T, van Cappellen van Walsum AM, Grotenhuis A. Tractography demonstrates dentate-rubro-thalamic tract disruption in an adult with cerebellar mutism. Cerebellum. 2013. 12: 617-22
35. Van Calenbergh F, Van de Laar A, Plets C, Goffin J, Casaer P. Transient cerebellar mutism after posterior fossa surgery in children. Neurosurgery. 1995. 37: 894-8
36. van Dongen HR, Catsman-Berrevoets CE, van Mourik M. The syndrome of ‘cerebellar’ mutism and subsequent dysarthria. Neurology. 1994. 44: 2040-6
37. Vandeinse D, Hornyak JE. Linguistic and cognitive deficits associated with cerebellar mutism. Pediatr Rehabil. 1997. 1: 41-4
38. Wells EM, Khademian ZP, Walsh KS, Vezina G, Sposto R, Keating RF. Postoperative cerebellar mutism syndrome following treatment of medulloblastoma: Neuroradiographic features and origin. J Neurosurg Pediatr. 2010. 5: 329-34
39. Wells EM, Walsh KS, Khademian ZP, Keating RF, Packer RJ. The cerebellar mutism syndrome and its relation to cerebellar cognitive function and the cerebellar cognitive affective disorder. Dev Disabil Res Rev. 2008. 14: 221-8
40. Zaheer SN, Wood M. Experiences with the telovelar approach to fourth ventricular tumors in children. Pediatr Neurosurg. 2010. 46: 340-3